Clear Sky Science · ru
Энергоэффективные научные вычисления с использованием химических резервуаров
Почему преобразование химии в вычисления важно
Современные суперкомпьютеры потребляют огромное количество электроэнергии для моделирования климата, разработки новых лекарств или обучения искусственного интеллекта. По мере приближения к физическим пределам традиционных чипов получение большей производительности на ватт становится всё сложнее и дороже. В этой статье рассматривается радикально иной путь: использование реальных химических реакций в качестве двигателя научных вычислений. Рассматривая молекулы и их взаимодействия как движущиеся части компьютера, авторы описывают, как будущие устройства могли бы решать сложные уравнения с гораздо меньшим энергопотреблением, чем современное цифровое оборудование.
От живых клеток к химическим калькуляторам
Живые клетки — мастера в решении задач. Они постоянно управляют тысячами реакций, чтобы адаптироваться, расти и выживать, при этом потребляя удивительно мало энергии. В основе этого поведения лежат сети химических реакций — взаимосвязанные реакции, скорости и концентрации которых меняются во времени. Эти сети можно описать обыкновенными дифференциальными уравнениями, тем же математическим языком, который используется для моделирования всего — от эпидемий до турбулентных течений. Основная идея работы в том, что раз химия уже следует этим уравнениям, её можно непосредственно использовать для выполнения вычислений, которые сегодня выполняют на кремниевых чипах.
Как уравнения превращаются в сети реакций
Авторы представляют ChemComp — программную платформу, которая принимает систему дифференциальных уравнений и систематически преобразует её в абстрактную сеть реакций. ChemComp использует современные технологии компиляции, чтобы разложить математическую задачу на шаблоны, которые можно представить идеализированными реакциями, а затем организует их в сеть с чётко определёнными видами веществ, связями и скоростями. Эти абстрактные реакции ещё не соответствуют реальным молекулам, но образуют проект химического компьютера. Далее платформа может искать в биохимических базах данных реальные мотивы реакций, которые ведут себя похоже, отдавая предпочтение практичным, безопасным и потенциально энергоэффективным вариантам для лабораторного воплощения.
Делегирование тяжёлой работы химическому резервуару
Для проверки идеи команда сосредоточилась на стиле машинного обучения, называемом резервуарными вычислениями. Здесь фиксированная динамическая система преобразует входной сигнал в богатый, запутанный узор внутренней активности, и обучается лишь простая считывающая прослойка, чтобы получить желаемый выход. В версии ChemComp резервуар — это набор реакций в хорошо перемешанном сосуде; меняющиеся концентрации химикатов формируют внутренние состояния. Авторы компилируют классическую двухпеременную систему, известную как модель Селькова–Шнакенберга — изначально использовавшуюся для изучения метаболических колебаний — в кандидатные сети реакций. Затем они имитируют, как эти сети реагируют во времени при подаче потоков веществ в сосуд и из него, и используют простую линейную регрессию, чтобы комбинировать траектории концентраций в приближение целевого решения.
Тестирование простых и более сложных химических сетей
Исследователи сравнивают два кандидатных резервуара: один с двумя химическими видами и двумя реакциями, и другой с пятью видами и пятью реакциями. Обе сети получают подходящие начальные концентрации и скорости потока, после чего моделируются во время работы. Даже меньшая система примерно воспроизводит колебательное поведение целевых уравнений, но более крупная сеть работает заметно лучше, уменьшая ошибку как при обучении, так и при тестировании. Сканируя разные начальные концентрации и константы скоростей реакций, авторы картируют области, где химическая система наиболее близка к желаемой динамике. Каждая реакция фактически играет роль базисной функции в задаче аппроксимации: чем разнообразнее доступные реакции, тем легче приблизить сложное поведение, но это увеличивает сложность системы.
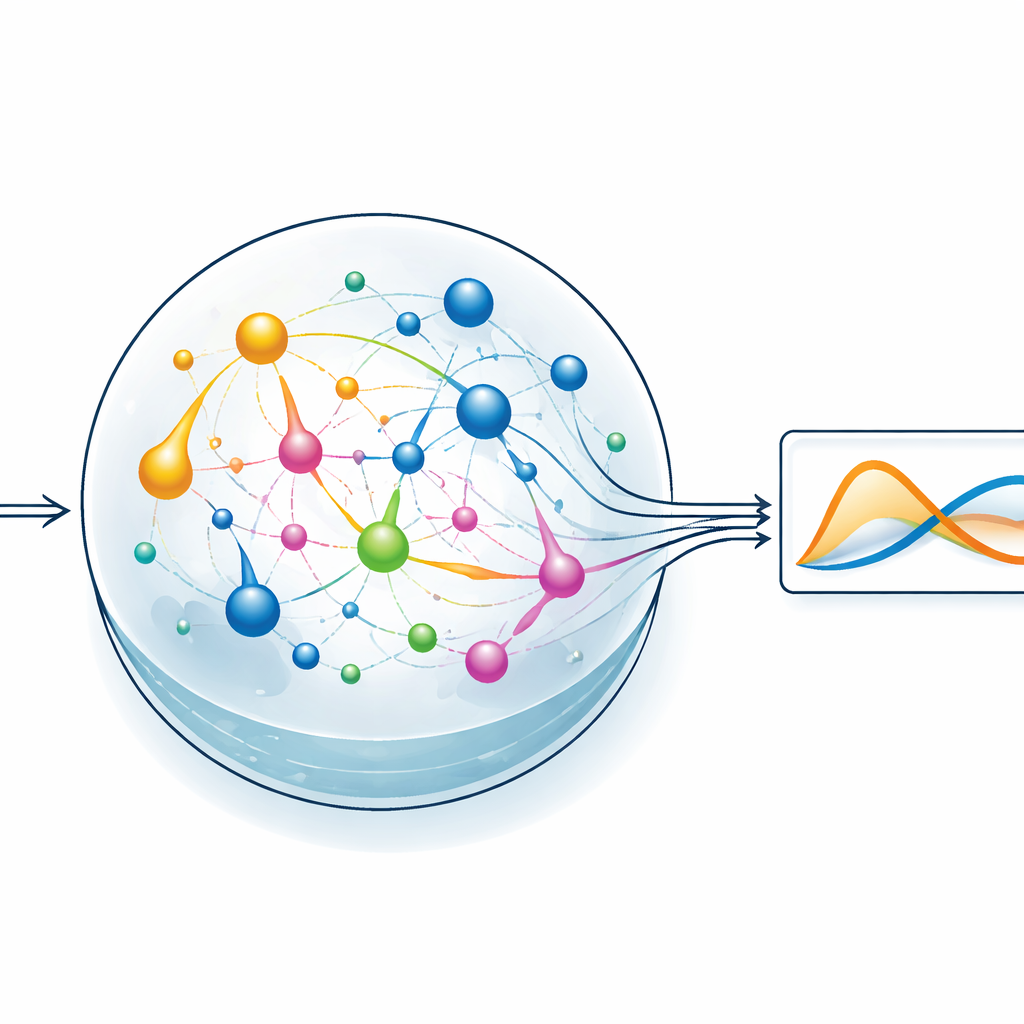
Путь к низкоэнергетичным вычислениям в лаборатории
Помимо моделирования, статья заглядывает в практические устройства. Обсуждается, как выбор реакций должен учитывать баланс энергопотребления, управляемость с помощью ферментов или катализаторов и возможность измерять ключевые вещества в реальном времени, например оптическими или электрохимическими методами. Авторы предполагают, что будущие микрофлюидные платформы смогут размещать тщательно подобранные сети реакций с пространственным контролем входов и встроенной системой датчиков. Хотя остаётся много инженерных задач — от преобразования уравнений в реальную химию до работы с шумом и пределов измерений — исследование показывает, что умеренные системы реакций уже способны эмулировать решения связанных дифференциальных уравнений. Для неспециалиста главный вывод таков: сама химия может служить аналоговым компьютером, открывая путь к научным вычислениям, опирающимся на энергоэффективные процессы, которые природа совершенствует миллиарды лет.
Цитирование: Johnson, C.G.M., Bohm Agostini, N., Cannon, W.R. et al. Energy-efficient scientific computing using chemical reservoirs. npj Unconv. Comput. 3, 17 (2026). https://doi.org/10.1038/s44335-026-00053-9
Ключевые слова: химические вычисления, энергоэффективные вычисления, резервуарные вычисления, сети химических реакций, обыкновенные дифференциальные уравнения