Clear Sky Science · ru
Комплексная платформа для предсказания реакций в гетерогенном катализе
Почему важно ускорять разработку катализаторов
Современное общество опирается на катализаторы при производстве топлива, пластмасс, удобрений и множества повседневных продуктов. Тем не менее поиск более эффективных катализаторов часто похож на поиск иголки в стоге сена: каждый материал способен одновременно способствовать тысячам микроскопических реакций. В этой статье представлен CARE — новая вычислительная платформа, которая с помощью правил и методов машинного обучения строит и моделирует запутанные сети реакций гораздо быстрее и полнее, чем прежде. Это обещает ускорить разработку чистых энергетических технологий и более эффективных химических процессов при значительном сокращении вычислительных затрат.
Распутывание переполненных путей реакций
На поверхности твердого катализатора молекулы не следуют по единому аккуратному пути от реагента к продукту. Они проходят через лабиринт краткоживущих интермедиатов и конкурирующих путей. Традиционные вычислительные методы опираются на человеческую интуицию, чтобы выбрать ограниченный набор возможных шагов, затем используют квантово-механические расчеты для оценки их энергий. Это работает для небольших сетей, но быстро теряет эффективность по мере усложнения системы, упуская редкие маршруты, которые могут определять долговременную активность, деактивацию или селективность. CARE решает эту проблему, автоматически строя очень большие сети реакций из простых правил-строителей, гарантируя включение всех правдоподобных событий разрыва и образования связей между углеродом, водородом и кислородом, даже тех, которые химики обычно могли бы отбросить.
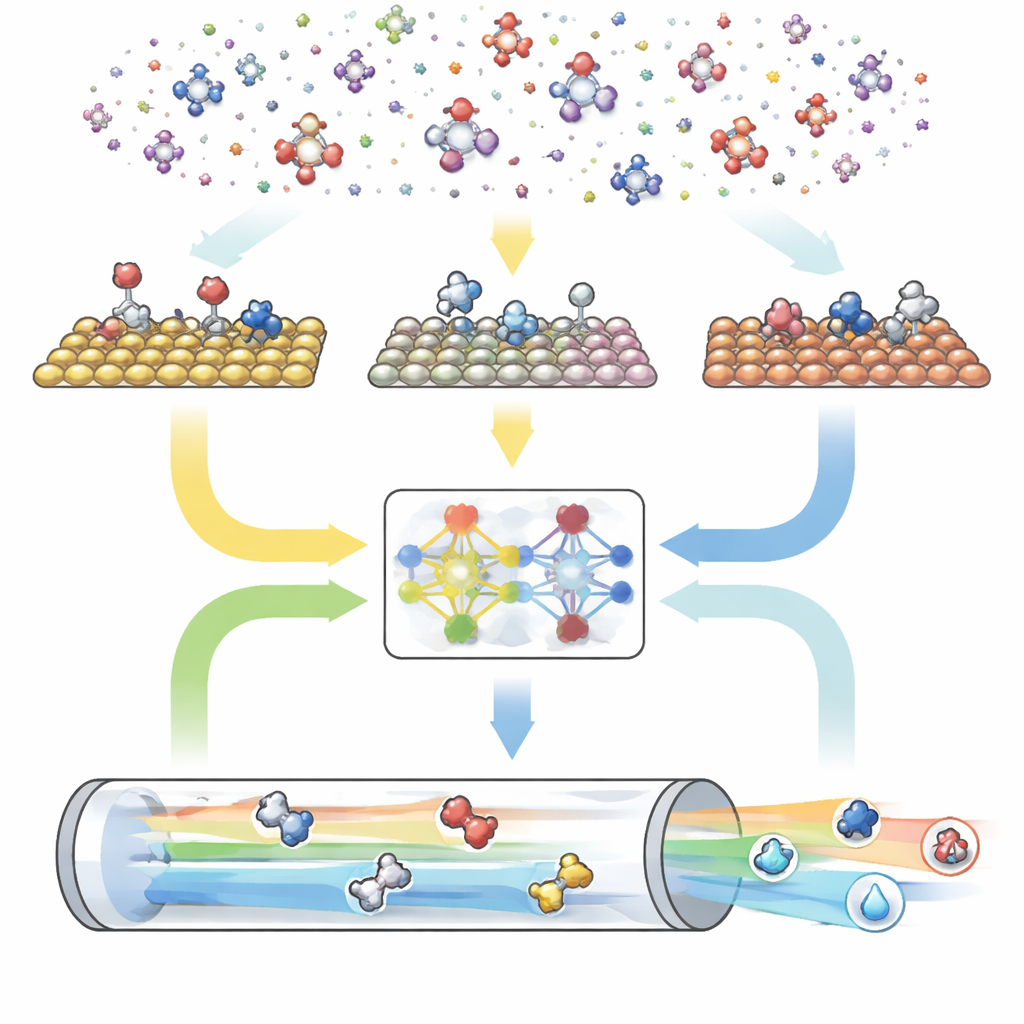
Цифровой двигатель из трёх частей
CARE собран как сквозной конвейер из трёх основных модулей. Во‑первых, генератор на основе правил определяет «химическое пространство», задавая максимальное число атомов углерода и кислорода и применяя простые шаблоны для создания всех подходящих молекул и их поверхностных форм. Во‑вторых, модуль оценки энергий использует современные модели машинного обучения — в частности графовую нейронную сеть GAME-Net-UQ — чтобы оценить энергии интермедиатов и переходных состояний на различных металлических поверхностях. Эта модель представляет структуру как сеть атомов и связей, возвращает и энергию, и неопределённость оценки, и обеспечивает точность в пределах нескольких десятых электронвольта при малой вычислительной нагрузке и высокой скорости. В‑третьих, микрокинетический решатель использует эти энергии, чтобы вычислить совместное протекание всех реакций в реалистичных условиях температуры, давления, напряжения и pH, прогнозируя общие скорости реакций, покрытие поверхности и селективность продуктов.
Проверка на реальных задачах: топливные молекулы и климатическая химия
Чтобы показать, что CARE — не просто теоретическая разработка, авторы применяют её к трём промышленно значимым задачам возрастающей сложности. Для разложения метанола — реакции важной для хранения водорода — они генерируют относительно небольшую сеть и оценивают её на множестве металлических катализаторов и кристаллических граней. CARE воспроизводит типичную «вулканическую» зависимость активности и правильно идентифицирует рутений как одного из лучших исполнителей, что согласуется с экспериментами, при этом требуя лишь крошечной доли вычислительного времени по сравнению с полными квантовыми расчётами. Далее рассматривают электрохимическое восстановление диоксида углерода на меди, с акцентом на образование C3-продуктов, таких как 1-пропанол и пропилен. Включив специальные шаги, учитывающие протоны, электроны и условия раствора, CARE захватывает влияние pH и приложенного напряжения на смещение путей и правильно предсказывает, что 1‑пропанол предпочтительнее пропилена, в согласии с детальными предыдущими исследованиями.
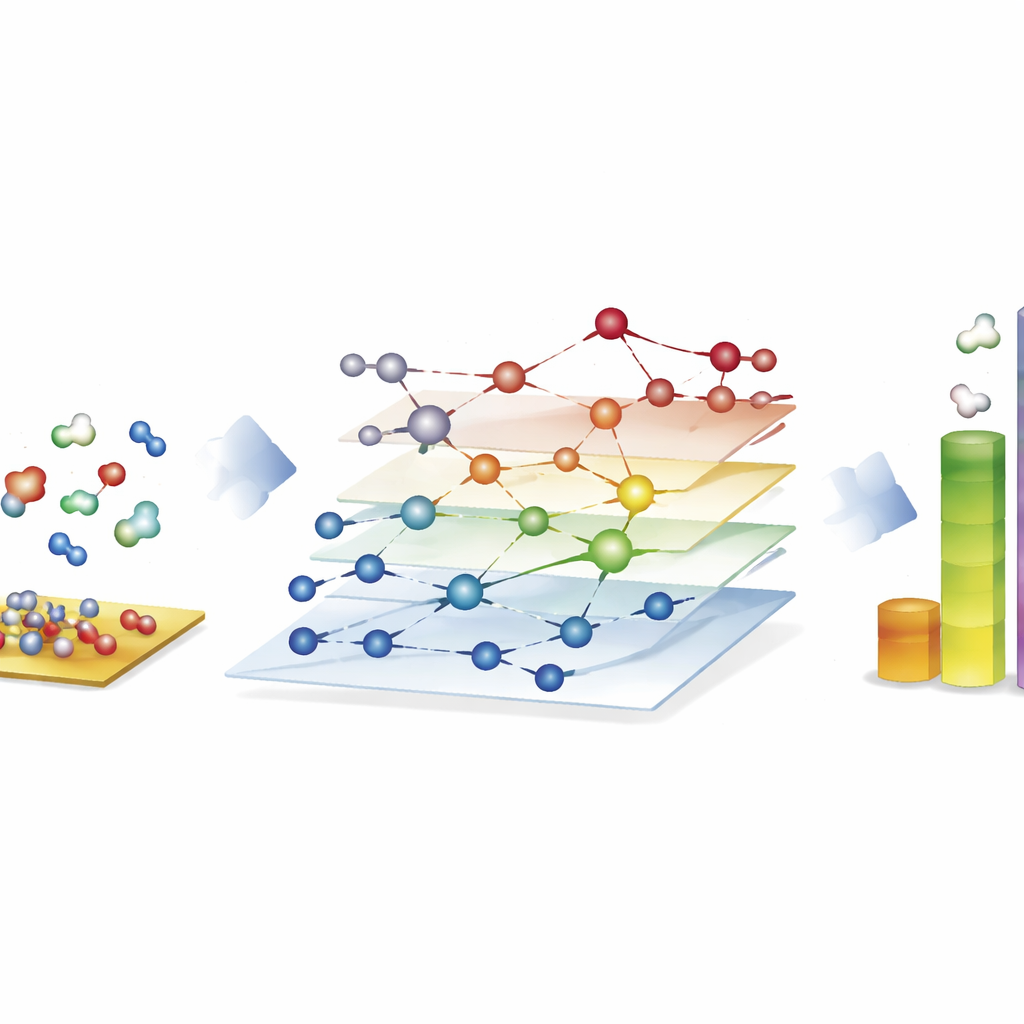
Исследование огромных сетей реакций для синтетического топлива
Самое впечатляющее демонстрируется на процессе Фишера–Тропша, который превращает смеси монооксида углерода и водорода в длинноцепочечные углеводороды для топлива и химикатов. Здесь авторы строят сети с почти 40 000 поверхностных видов и около 370 000 элементарных реакций — далеко за пределами того, что традиционные квантово‑ориентированные исследования могут полноценно исследовать. С помощью CARE они оценивают все интермедиаты и ключевые барьеры реакций на поверхностях кобальта, железа, никеля и рутения всего за несколько часов на стандартном оборудовании, что быстрее примерно в миллион раз по сравнению с прямыми квантовыми расчётами. Микрокинетические симуляции этих сетей воспроизводят известные тенденции: кобальт и железо предпочитают образовывать более длинные цепи углеводородов, железо даёт больше диоксида углерода через побочные реакции, а никель склонен к более сильной гидрогенизации. Хотя некоторые детали, например выход метана, остаются несовершенными, платформа выявляет, какие шаги образования связей доминируют в росте цепей, и подчёркивает области, требующие доработки моделей.
Что это значит для будущих катализаторов
Для неспециалистов ключевой посыл таков: CARE предоставляет практический способ исследовать огромные реакционные пространства на каталитических поверхностях, которые ранее были недоступны. Автоматизируя генерацию сетей, подключая быстрые «суррогатные» модели машинного обучения для квантовых энергий и эффективно решая получающиеся кинетические уравнения, система может ранжировать кандидат‑катализаторы, выявлять перспективные условия работы и обнаруживать неожиданные пути с гораздо меньшим человеческим уклоном и вычислительными затратами. Авторы отмечают оставшиеся проблемы — например лучшее учёт плотных покрытий поверхностей, влияние растворителя и ещё более крупные сети — но работа указывает на будущее, в котором компьютеры смогут быстро скринировать сложные реакции, от восстановления CO2 до переработки пластика и обновления биомассы, направляя эксперименты к наиболее перспективным идеям вместо опоры на метод „метод проб и ошибок".
Цитирование: Morandi, S., Loveday, O., Renningholtz, T. et al. An end-to-end framework for reactivity in heterogeneous catalysis. Nat Chem Eng 3, 169–180 (2026). https://doi.org/10.1038/s44286-026-00361-8
Ключевые слова: гетерогенный катализ, сети реакций, машинное обучение, микрокинетическое моделирование, синтез Фишера–Тропша