Clear Sky Science · ru
Гематопоэтическая экспрессия cIAP2 приводит к воспалению и сердечной недостаточности после инфаркта миокарда
Почему важно укротить воспаление после сердечного приступа
Пережить сердечный приступ — это только начало. В последующие дни и недели иммунная система устремляется к повреждённой ткани, чтобы убрать разрушенные клетки и начать восстановление. Но если воспаление слишком острое или длительное, оно может трансформировать полезный процесс заживления в источник постоянного повреждения сердца и развития сердечной недостаточности. В этом исследовании обнаружен ключевой молекулярный переключатель в кроветворных иммунных клетках, который поддерживает этот воспалительный «огонь», и показано, что его отключение может защитить сердце в экспериментальных моделях.
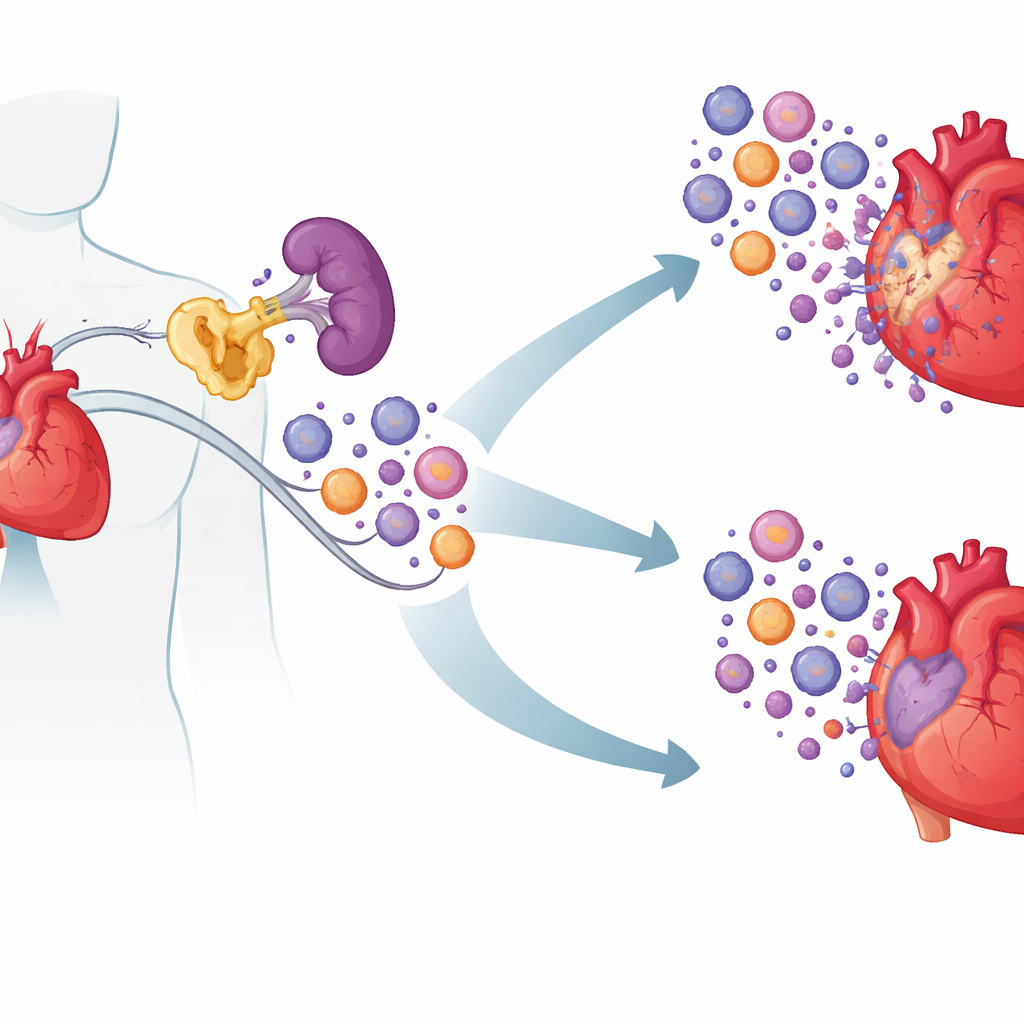
Скрытый виновник внутри иммунных клеток
Исследователи сосредоточились на белке cIAP2, прежде всего известном как фактор, помогающий клеткам рака избегать гибели. Анализ крови пациентов с острыми сердечными проблемами показал, что уровни cIAP2 были выше у людей с недавним инфарктом и при ишемической сердечной недостаточности по сравнению со здоровыми донорами или пациентами со стабильной коронарной болезнью. Ткани сердца человека и мышей демонстрировали ту же картину: cIAP2 резко возрастал вскоре после инфаркта, тогда как его родственный белок cIAP1 — нет. При анализе существующих наборов данных экспрессии генов команда обнаружила, что повышение cIAP2 происходило синхронно с генами, связанными с агрессивными миелоидными воспалительными клетками, что наводит на мысль о том, что cIAP2 может усиливать иммунный ответ после инфаркта, а не просто отражать степень повреждения.
Меньше cIAP2 — меньше повреждений сердца
Чтобы проверить причинно-следственную связь, учёные сравнили обычных мышей и животных, генетически лишённых cIAP2. После экспериментального инфаркта у мышей без cIAP2 наблюдались меньшие рубцы, лучшая сократительная функция и меньшее накопление жидкости в лёгких — все признаки более сохранённого сердца. Профит отмечался как у самцов, так и у самок. Микроскопия показала меньше гибнущих кардиомиоцитов в критических переходных зонах, а молекулярные анализы — более низкие уровни маркеров стресса и ремоделирования через несколько недель. Напротив, удаление cIAP1 не давало такой защиты и в некоторых условиях даже могло ухудшать исход, что указывает на уникальную вредную роль cIAP2 в этом контексте.
Роль селезёнки как воспалительного резервуара
Ключевым оказался вопрос локации действия cIAP2. Пересадка костного мозга между нормальными и лишёнными cIAP2 мышами показала, что именно cIAP2 в кроветворных (гематопоэтических) клетках обусловливал большую часть повреждений. Если иммунные клетки лишены cIAP2, а остальной организм нормален, сердце оказывается лучше защищено; обратная пересадка усиливала вред. При подробном изучении иммунных органов выяснилось, что после инфаркта селезёнка выступает в роли резервуара, генерирующего миелоидные клетки — нейтрофилы, воспалительные моноциты и дендритные клетки — которые затем направляются в сердце. У мышей без cIAP2 число таких селезёночных миелоидных клеток было ниже и они были более склонны к гибели, тогда как лимфоциты оставались в основном неизменными. Сигналы, связанные с воспалительными путями, были ослаблены, что говорит о том, что cIAP2 обычно помогает миелоидным клеткам выживать и продолжать отвечать на сигналы опасности.
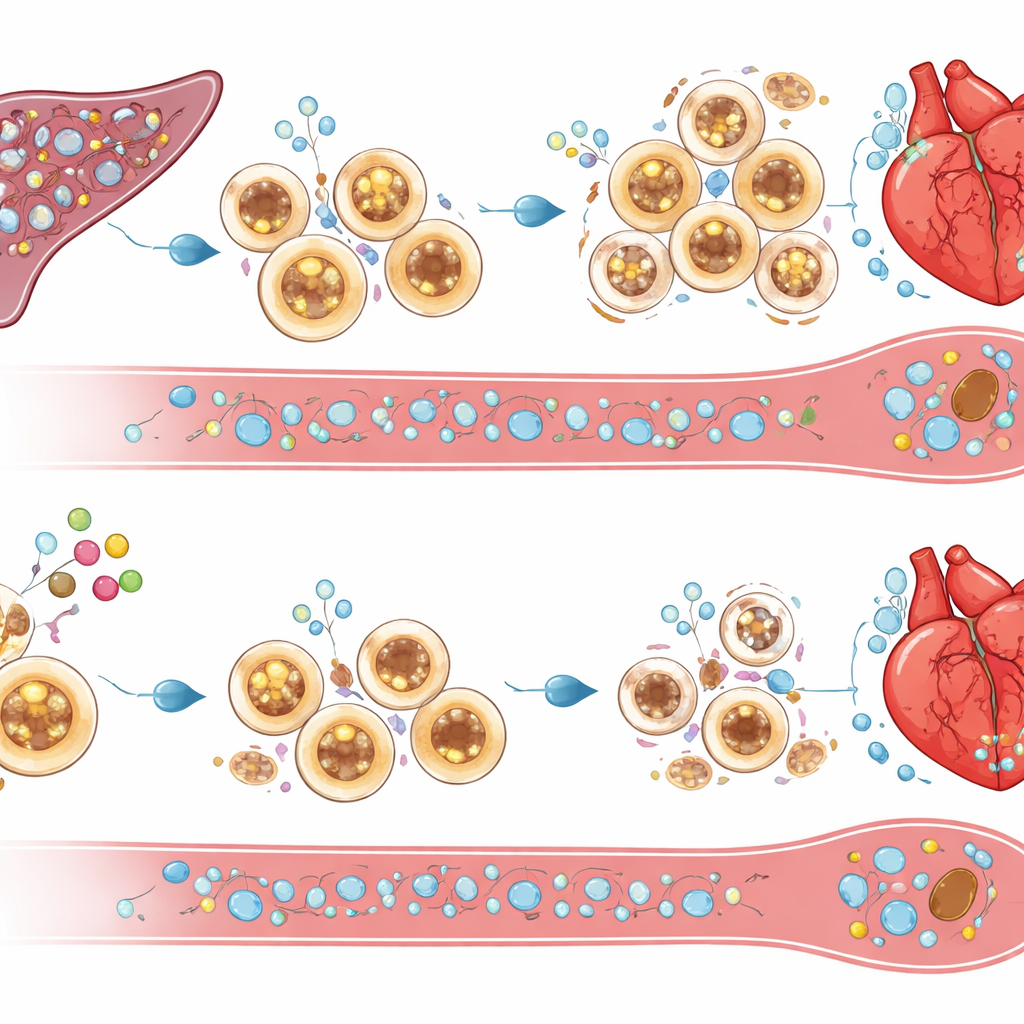
Преобразование сигналов выживания в самограничивающуюся очистку
Что же убивает избыточные воспалительные клетки при отсутствии cIAP2? Исследование указывает на молекулы, индуцирующие гибель, такие как TRAIL и его рецептор DR5, а также на сигналы, связанные с TNF, которые были повышены в селезёнках и костном мозге мышей без cIAP2 после инфаркта. Экспериментальная блокада TRAIL спасала клетки селезёнки от гибели, восстанавливала массовую инфильтрацию сердца иммунными клетками и устраняла функциональные преимущества отсутствия cIAP2. Это позволяет предположить, что cIAP2 обычно защищает миелоидные клетки от этих сигналов смерти, позволяя им накапливаться и продлевать воспаление. Без cIAP2 те же сигналы «подрезают» селезёночный резерв, сокращая приток агрессивных клеток, которые в противном случае затопили бы повреждённое сердце.
Ориентация на переключатель для будущих терапий
Важно, что команда показала возможность фармакологического воздействия на этот путь с помощью уже известных малых молекул класса Smac-миметиков, изучаемых в онкологии. Применение соединения LCL161 выборочно вызвало распад белков cIAP в селезёночных иммунных клетках вскоре после инфаркта, не выводя при этом защитные белки из самой сердечной ткани. У обработанных мышей было меньше циркулирующих воспалительных клеток, меньшие рубцы, лучшая функция сердца и повышенная выживаемость по сравнению с необработанными животными. Однократная низкая доза, введённая на следующий день после инфаркта, была достаточна для индуцирования контролируемой гибели селезёночных миелоидных клеток, повышения локального уровня TRAIL и снижения воспаления в сердце, при этом общее число иммунных клеток восстановилось к четырём неделям. В совокупности эти результаты выделяют cIAP2 как центральный фактор выживания для воспалительных клеток после повреждения сердца и позволяют предположить, что кратковременная целевая ингибиция cIAP2 может стать новым иммунотерапевтическим подходом к предотвращению сердечной недостаточности после инфаркта миокарда.
Цитирование: Smyth, D., Zhang, L., Al-Khalaf, M. et al. Hematopoietic expression of cIAP2 drives inflammation and heart failure after myocardial infarction. Nat Cardiovasc Res 5, 246–261 (2026). https://doi.org/10.1038/s44161-026-00782-x
Ключевые слова: инфаркт миокарда, воспаление, иммунные клетки, сердечная недостаточность, Smac-миметик