Clear Sky Science · ru
Беспрецедентная устойчивость физически информированных моделей атомной энергии при комнатной температуре и выше
Почему это важно для повседневной химии
Компьютерные симуляции — это рабочая лошадка современной химии и материаловедения. Они позволяют учёным наблюдать, как молекулы вращаются, вибрируют и сталкиваются в виртуальной среде вместо дорогих и длительных экспериментов. Но когда эти симуляции зависят от машинного обучения, они могут внезапно «взрываться», порождая невозможные молекулярные формы — особенно при повышенных температурах. В этой работе представлен новый тип физически информированной модели машинного обучения, который способен запускать такие симуляции на чрезвычайно длительное время при температурах до 1000 К, не разрушаясь.
От хитрых упрощений к хрупким симуляциям
Традиционная квантовая химия вычисляет молекулярные энергии с высокой точностью, но крайне медленно. Более простые поля сил быстры, но часто приближённы. Потенциалы, обученные методом машинного обучения, стремятся соединить лучшее из обоих миров: они изучают «короткий путь» от геометрии молекулы до энергии и сил, а затем используют этот путь для молекулярной динамики. На бумаге многие такие модели выглядят превосходно, демонстрируя крошечные средние ошибки на стандартных тестовых наборах. На практике эти цифры могут вводить в заблуждение. Когда молекулы во время симуляции исследуют новые конфигурации — особенно при высоких температурах — многие модели выходят за пределы областей, на которых их обучали. Вместо того чтобы мягко вернуть молекулы к реалистичным формам, они могут предсказывать силы, которые растягивают или раздавливают связи до тех пор, пока система не станет нефизической и симуляция не рухнет.
Построение моделей на квантово-обусловленных блоках
Авторы решают эту хрупкость, изменив то, чему модель учится, и как её направляет предшествующая физика. Они используют фреймворк под названием FFLUX, основанный на подходе Interacting Quantum Atoms (IQA). В IQA молекула делится на «топологические атомы», энергия каждого из которых определяется непосредственно из квантовой механики. Эти атомные энергии имеют физический смысл и суммируются в общую энергию молекулы. Вместо обучения произвольным локальным энергиям новые модели на основе гауссовских процессов изучают именно эти квантово-обусловленные атомные энергии, обеспечивая глубокую физическую опору для каждого предсказания. В качестве сложных учебных примеров использовали четыре гибкие органические молекулы — глицин и серин с пептидной защитой, малондальдегид и аспирин — из‑за их многочисленных внутренних движений и известной трудности для существующих моделей на базе машинного обучения.
Обучение модели ожидать проблем
Ключевая новация заключается в том, как гауссовский процесс настраивается ещё до того, как увидит данные: его «средняя функция», которая кодирует предположения модели в плохо изученных областях. В большинстве предыдущих работ эту среднюю функцию просто задавали равной нулю, фактически притворяясь, что у модели нет априорных ожиданий. Авторы вместо этого намеренно сдвигают эту среднюю функцию в сторону более высоких значений атомных энергий, сохраняя при этом физическую правдоподобность. Такое проектное решение означает, что когда модель вынуждена экстраполировать — например, при временном чрезмерном растяжении связей — она естественно предпочитает предсказания, штрафующие экстремальные искажения. В обширных тестах версии модели, различавшиеся только этой априорной настройкой, вели себя очень по‑разному. Модели с традиционной или низкоэнергетической средней функцией зачастую не выдерживали и пикосекунды, прежде чем молекула «взорвалась» или «вдавилась». Напротив, лучшая средняя функция с высоким уровнем энергии (названная MF5) дала симуляции, сохранявшиеся стабильными на протяжении всего наносекундного тестового окна при температурах от 300 до 1000 К для всех четырёх молекул.
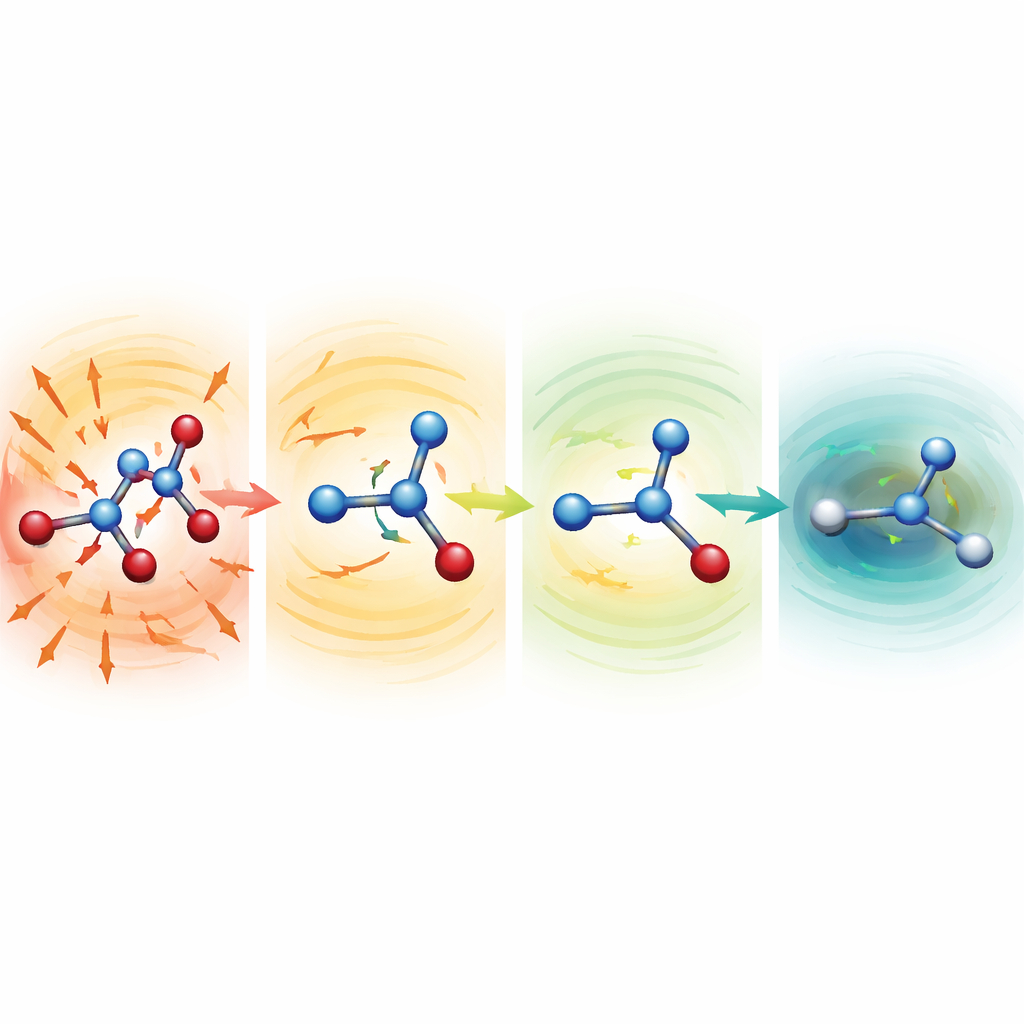
Наблюдая, как искажённые молекулы исцеляются
Чтобы понять, почему устойчивые модели работают так хорошо, исследователи запускали симуляции с намеренно повреждёнными структурами с сильно растянутыми или сжатыми связями. Для серина, аспирина и малондальдегида эти начальные конфигурации лежали на сотни и даже более тысячи килокалорий на моль выше нормальной структуры — состояния, которые обычно приводят к катастрофе. При более «мягких» средних функциях молекулы быстро разлетались. Однако в настройке MF5 предсказанные силы сразу указывали в направлении, сокращающем удлинённые связи и удлиняющем сжатые. В течение десятков-сотен шагов времени молекулы расслаблялись до реалистичных форм и затем продолжали стабильно эволюционировать. Команда также показала, что, не обучаясь на силах, те же модели могут направлять оптимизации геометрии дипептида аланина, воспроизводя известные низкоэнергетические конформации и относительные энергии с точностью в несколько десятых килокалории на моль, но при примерно в 200 раз меньших вычислительных затратах по сравнению с полными квантовыми расчётами.
Долгие горячие симуляции на обычном оборудовании
Устойчивость — это не только пережить один тяжёлый старт; это выдерживать миллионы или миллиарды шагов времени. Авторы подвергли свои лучшие модели дальнейшей проверке, запустив 50 независимых симуляций при 500 К, каждая по 10 наносекунд, для каждой из четырёх тестовых молекул. Ни одна из этих прогонов не завершилась аварией, в сумме давая полмикросекунды симуляционного времени — необычный результат для передовых полей сил, обученных машинным методом. Ещё более примечательно, что симуляции выполнялись эффективно на стандартных CPU, шаг в шаг соперничая или опережая некоторые заметные нейронные потенциалы, для которых требуются мощные GPU. На протяжении всего времени молекулы исследовали богатый набор форм и метастабильных состояний, что показывает: устойчивость достигнута не путём искусственного «замораживания» движений или навязывания жёстких структур.
Что это означает для будущего молекулярного моделирования
Для неспециалистов ключевое сообщение таково: не все модели машинного обучения с низкой средней ошибкой заслуживают доверия при сильной нагрузке. Основав свои модели на квантово-выведенных атомных энергиях и тщательно настроив априорные ожидания модели в сторону высокоэнергетических состояний, авторы создали семейство потенциалов, которые естественным образом генерируют «восстанавливающие силы» — молекулярный эквивалент страховочного ремня — поддерживающие физичность симуляций даже при высоких температурах и при искажённых начальных структурах. Этот подход обещает более надёжные и долгие симуляции сложных молекул и указывает путь к будущим расширениям, где похожие физически информированные модели будут работать в конденсированных фазах и учитывать тонкие взаимодействия, такие как дисперсия, при сохранении вычислительной практичности.
Цитирование: Isamura, B.K., Aten, O., Nosratjoo, M. et al. Unprecedented robustness of physics-informed atomic energy models at and beyond room temperature. Commun Chem 9, 138 (2026). https://doi.org/10.1038/s42004-026-01965-0
Ключевые слова: поля сил на основе машинного обучения, устойчивость молекулярной динамики, потенциалы на основе гауссовских процессов, физически информированное моделирование, квантовые атомные энергии