Clear Sky Science · ru
Деметилаза гистонов KDM7A негативно регулирует фибротическую поляризацию макрофагов и прогрессирование легочного фиброза
Почему рубцевание лёгких важно для всех
Когда в лёгких развиваются стойкие рубцы, дыхание становится ежедневной борьбой. Это состояние, известное как лёгочный фиброз, поражает миллионы людей и на сегодняшний день не излечимо — существуют лишь препараты, замедляющие повреждение. В этом исследовании учёные обнаружили ранее скрытый молекулярный «тормоз» внутри иммунных клеток, называемых макрофагами, который помогает сдерживать образование рубцов в лёгких. Понимание этого механизма может открыть путь к новым методам лечения не только лёгочного фиброза, но и других заболеваний, где вредное рубцевание и неконтролируемое воспаление идут рука об руку.
История клеток‑хамелеонов иммунной системы
Макрофаги — клетки передовой защиты, патрулирующие ткани, удаляющие мусор и способствующие восстановлению повреждений. Но они ещё и «превращаются»: в одних ситуациях они становятся провоспалительными бойцами, в других — заживляющими клетками, которые могут стимулировать образование рубцов. Особенно связанный с фиброзом тип — профибротические макрофаги (Fib‑Mac) — тесно ассоциируется с лёгочным фиброзом. Эти клетки вырабатывают молекулы, активирующие фибробласты, которые затем откладывают избыток коллагена и других компонентов матрикса, постепенно уплотняя лёгкое. Авторы хотели понять, какие внутригенетические «настройки» в макрофагах определяют, станут ли они опасными Fib‑Mac или сохранят более сбалансированное, защитное состояние.

Эпигенетический тормоз, скрытый в геноме
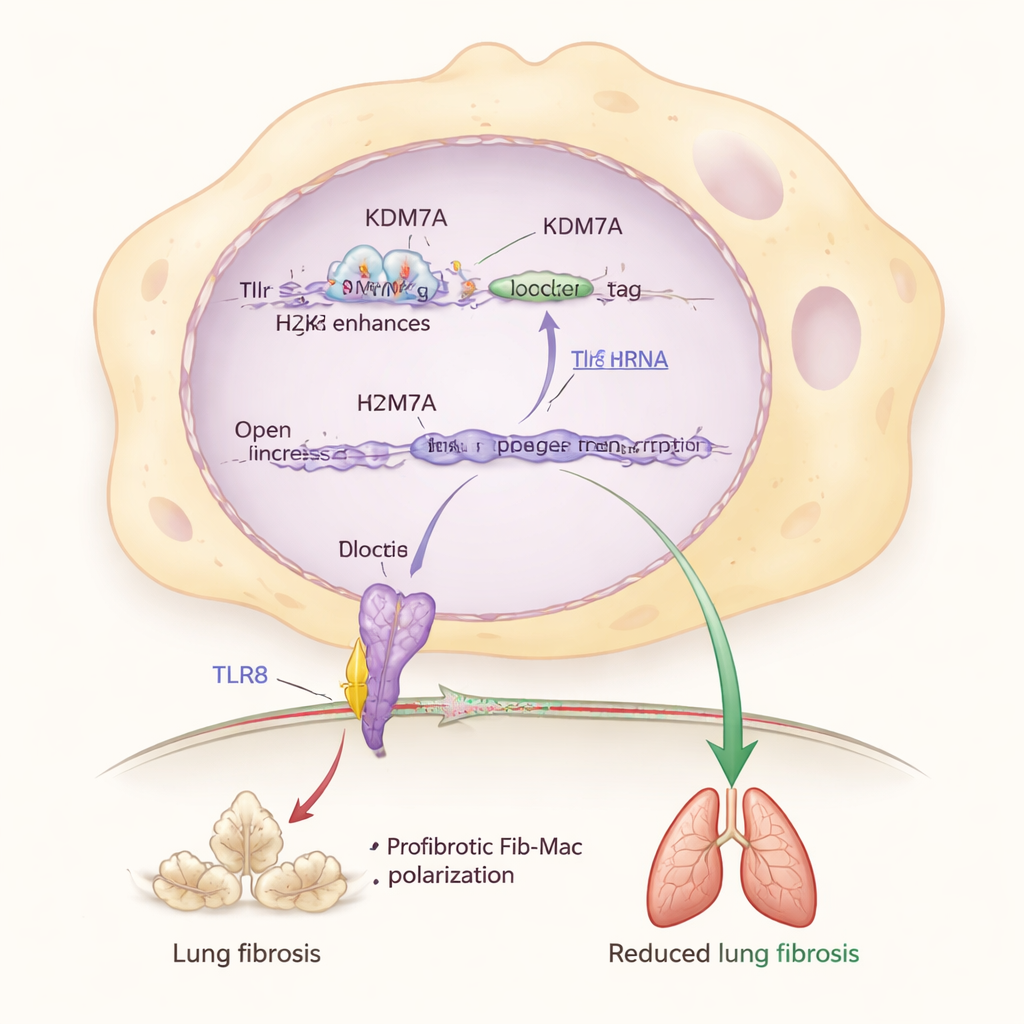
Группа начала со скрининга сотен известных эпигенетических регуляторов — белков, которые тонко настраивают упаковку ДНК и включают или выключают гены. С помощью РНК‑секвенирования в человеческих и мышиных макрофагах они обнаружили, что фермент KDM7A сильно активируется, когда макрофаги направляются к фибротическому, заживляющему состоянию. KDM7A — это деметилаза гистонов: она удаляет определённые химические метки с гистонов, вокруг которых намотана ДНК. Эта картина намекала, что KDM7A может действовать как встроенный сигнальный «тормоз», активируемый именно тогда, когда макрофаги начинают склоняться к фенотипу, стимулирующему рубцевание.
Чтобы проверить гипотезу, исследователи использовали мышей с удалённым геном Kdm7a и вызывали повреждение лёгких с помощью химотерапевтического препарата блеомицин — стандартной модели лёгочного фиброза. В ранние сроки после повреждения лёгочная ткань выглядела похоже у нормальных и Kdm7a‑дефицитных животных. Но через три недели у мышей без Kdm7a наблюдалось значительно более обширное рубцевание, спад мелких воздушных мешочков и более высокие «оценки Ашкрофта», количественно отражающие фиброз. Гены, участвующие в синтезе коллагена и других фиброзных путях, были более активны у этих нокаутов, что подтверждает: потеря Kdm7a делает лёгкие более уязвимыми к длительному повреждающему рубцеванию.
Как KDM7A отводит макрофаги от судьбы, приносящей рубцы
С помощью одно‑клеточного РНК‑секвенирования авторы внимательно рассмотрели отдельные клетки лёгких у повреждённых мышей. Они обнаружили, что при отсутствии Kdm7a в стромальном слое лёгкого существенно расширяется определённый подтип макрофагов и приобретает выраженный Fib‑Mac сигнатурный профиль, экспрессируя гены такие как Arg1, Spp1 и Trem2. Дальнейшие эксперименты в культурах макрофагов показали, что удаление Kdm7a усиливает маркеры Fib‑Mac и перестраивает метаболизм клеток в пользу путей, поддерживающих синтез коллагена и поддержанную активацию. Иначе говоря, KDM7A обычно сдерживает как генетические, так и метаболические программы, которые заставляют макрофагов переходить в состояние, стимулирующее фиброз.
Копнув глубже, исследователи выделили ключевого партнёра в этой системе торможения: сенсорный белок TLR8, который распознаёт фрагменты РНК внутри иммунных клеток. Они показали, что KDM7A поддерживает включённость гена Tlr8, удаляя репрессивную химическую метку (H3K27me2) с энхансера рядом с Tlr8. При инактивации Kdm7a эта метка накапливалась, уровень Tlr8 падал, а черты Fib‑Mac усиливались. Прямое уменьшение Tlr8 в макрофагах также сдвигало их в фибротическое состояние, тогда как активация или избыточное производство TLR8 возвращало их в прежнее состояние, даже при отсутствии Kdm7a. Это помещает путь KDM7A–TLR8 в центр молекулярной цепи, защищающей лёгкие от чрезмерного образования рубцов.
От стареющих лёгких к человеческим заболеваниям
Чтобы связать эти результаты с клинической практикой, команда изучила ткань лёгких пациентов с фибротическим заболеванием лёгких. По сравнению со здоровыми контрольными образцами в фибротических лёгких было гораздо больше макрофагов с маркерами Fib‑Mac, но в этих же клетках заметно снижались уровни KDM7A и TLR8. Повторный анализ доступных одно‑клеточных наборов данных пациентов с идиопатическим лёгочным фиброзом подтвердил эту закономерность: по мере усиления Fib‑Mac сигнатур экспрессия KDM7A падала. Исследователи также проанализировали большую мышиную атласную базу и обнаружили, что экспрессия Kdm7a и Tlr8 в макрофагах у самцов снижается с возрастом, что коррелирует с повышенным риском лёгочного фиброза у пожилых мужчин. Это указывает на то, что возраст‑ и пол‑зависимое ослабление тормоза KDM7A–TLR8 может частично объяснять, кто наиболее уязвим к тяжёлому рубцеванию лёгких.
Что это значит для будущих терапий
Проще говоря, работа показывает, что наша иммунная система несёт внутренний механизм безопасности, который не позволяет клеткам, участвующим в восстановлении, стать чрезмерно активными и превратиться в драйверов постоянного рубцевания. KDM7A, действуя через TLR8, удерживает макрофаги от застревания в профибротическом режиме и тем самым помогает поддерживать гибкую, функциональную ткань лёгких после повреждения. Когда эта система даёт сбой — из‑за генетической потери, старения или других факторов — макрофаги с большей вероятностью становятся «усилителями рубцов», усугубляя фиброз. Описав этот эпигенетический тормоз, исследование указывает на новые терапевтические направления: препараты, усиливающие активность KDM7A, имитирующие её эффекты или аккуратно стимулирующие TLR8, теоретически могут дополнять существующие антифибротические терапии и обеспечить лучшую защиту от прогрессирующего, ограничивающего жизнь рубцевания лёгких.
Цитирование: Funagura, N., Koga, T., Etoh, K. et al. Histone demethylase KDM7A negatively regulates fibrotic macrophage polarization and lung fibrosis progression. Commun Biol 9, 309 (2026). https://doi.org/10.1038/s42003-026-09610-1
Ключевые слова: легочный фиброз, макрофаги, эпигенетика, KDM7A, TLR8