Clear Sky Science · ru
haCCA: много-модульная интеграция спотовых пространственных транскриптомов и метаболомов
Почему важно картировать молекулы на месте
Наши тела состоят из множества крошечных «районов» клеток, у каждого из которых свой набор активных генов и химических веществ. До недавнего времени учёные изучали эти молекулы, измельчая ткани в однородную массу, и при этом теряли всю информацию о «где» они находятся. В этой статье представлен новый вычислительный метод под названием haCCA, который объединяет два мощных образных метода, чтобы исследователи могли видеть in situ, как гены и малые молекулы распределены по настоящим тканям и опухолям. Такая карта может выявить скрытые паттерны болезни и подсказать более точные стратегии лечения.
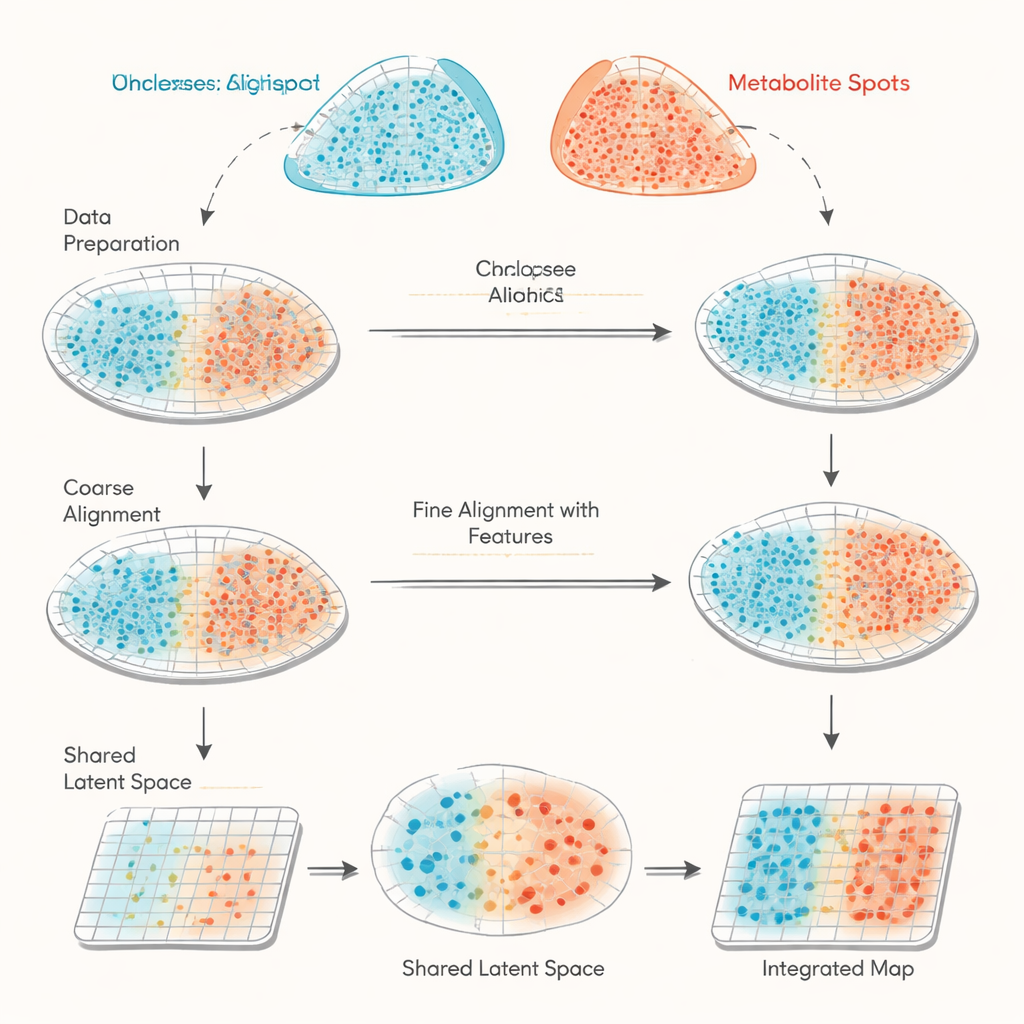
Два разных взгляда на одну и ту же ткань
Исследование сосредоточено на объединении данных из двух пространственных методов, которые всё чаще используются в биологии. Пространственная транскриптомика фиксирует, какие гены включены в тысячах маленьких спотов по срезу ткани. MALDI-масс-спектрометрическое визуализирование фиксирует количество многих малых молекул, таких как метаболиты и липиды, на аналогично плотных сетках спотов. Проблема в том, что эти два прибора не измеряют точно одинаковые позиции и не охватывают один и тот же набор признаков, поэтому их данные похожи на два несовмещённых плана с разными легендами. Существующие подходы в основном пытаются сопоставить форму срезов тканей только по координатам, что может быть неточно и лишено механизма проверки качества выравнивания.
Более умный способ выравнивания молекулярных карт
haCCA (сокращение от hierarchical anchor-guided canonical correlation analysis) решает эту задачу, сочетая геометрию и биологию. Сначала он выполняет двухэтапное «морфологическое выравнивание» сеток спотов от двух технологий. Эксперты вручную выбирают несколько совпадающих ориентиров на изображениях тканей, чтобы грубо скорректировать смещения и вращения, а затем автоматизированный этап уточняет выбивающиеся точки у разрывов или на участке с отсутствующими фрагментами. Далее метод ищет «якорные» пары спотов, которые близки по пространству и расположены в локально однородных областях, что делает их вероятными представителями одной и той же зоны ткани. На основе этих якорных спотов haCCA рассчитывает, какие гены и метаболиты склонны изменяться совместно, и сводит их в общее низкоразмерное представление, фиксирующее их сильнейшие совместные паттерны.
Преобразование корреляций в единое изображение ткани
Имея и пространственные координаты, и общее молекулярное представление, haCCA решает задачу оптимизации, чтобы оценить, насколько вероятно соответствие каждого генного спота каждому метаболитному споту. Этот шаг спроектирован так, чтобы сохранять пространственную близость спотов, но также учитывать их сходство в объединённом ген–метаболитном профиле. Итогом становится «план переноса», который связывает каждую точку одного набора данных с её наилучшим партнёром в другом, создавая интегрированную многомодальную карту. На тщательно смоделированных тестовых данных — где истинные связи известны — авторы показывают, что каждый этап рабочего процесса (грубое выравнивание, уточнённое выравнивание и сопоставление с учётом признаков) последовательно улучшает три независимые метрики точности. По сравнению с инструментами, опирающимися в основном на геометрию, haCCA стабильно достигает лучшего выравнивания и более достоверной передачи меток областей.
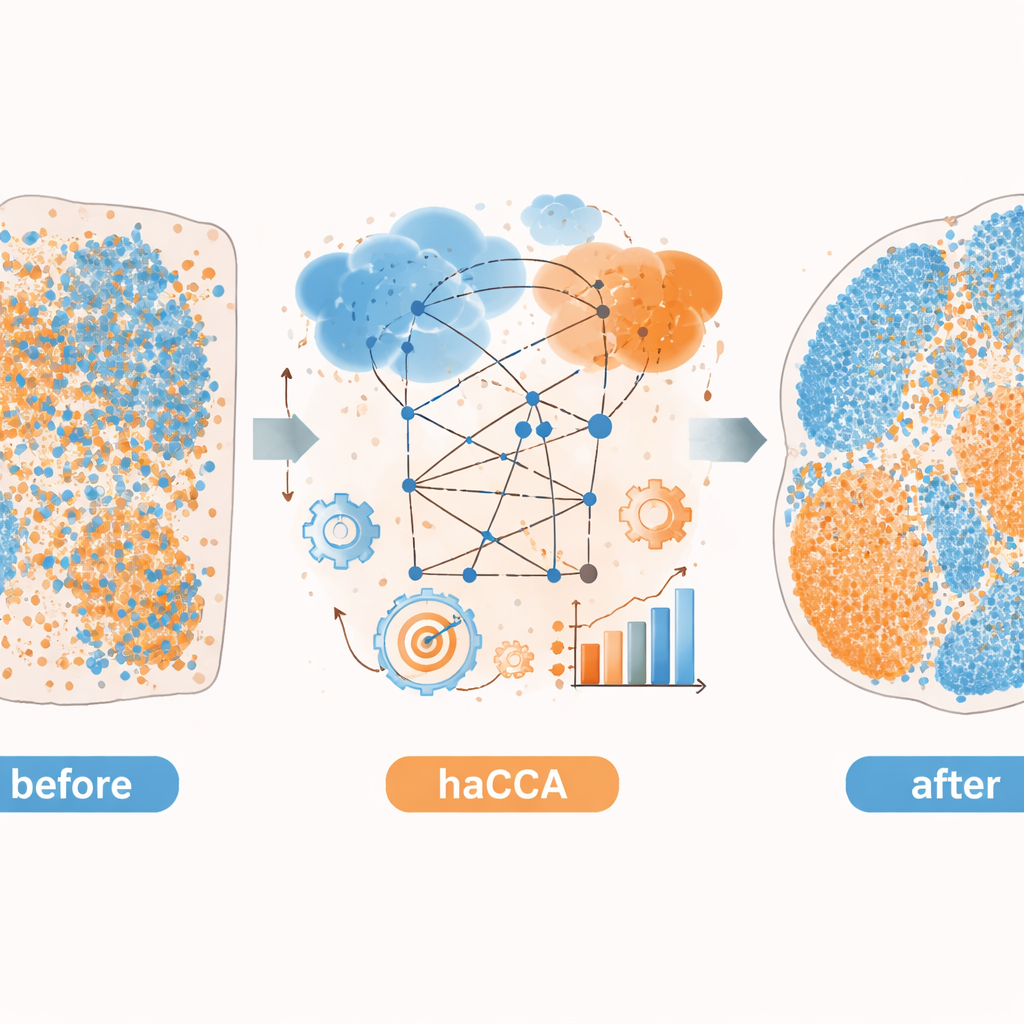
Выявление скрытой биологии в мозге и раке печени
Затем авторы применяют haCCA к реальным тканям мозга мыши и опухолям печени. Для мозга они интегрируют коммерческие данные пространственной транскриптомики с метаболитными изображениями с тех же или соседних срезов. Метод сохраняет известные метаболические территории и реконструирует ожидаемые наложения, такие как со-локализация дофамина с геном, кодирующим его ключевой фермент. Совместная кластеризация генов и метаболитов показывает, что объединённые данные разделяют тканевые подрегионы более тонко, чем любая модальность по отдельности. В доклинической модели внутрипечёночной холангиокарциномы, типа рака печени, они используют haCCA для сравнения опухолей, способных или неспособных формировать внеклеточные ловушки нейтрофилов — сетчатые структуры, выделяемые иммунными клетками. Интегрированные карты показывают, что при наличии таких ловушек ген Scd1 и связанные с ним жирные кислоты обогащены в злокачественных областях, указывая на сдвиг в сторону изменённого жирового метаболизма в опухоли.
Что это значит для будущих исследований
Проще говоря, haCCA похож на совмещение аэрофотоснимков, сделанных разными камерами — одна чувствительна к контурам зданий, другая к тепловым сигналам — чтобы получить более чёткое представление о том, что происходит в каждом городском квартале. Точное объединение мест активности генов и мест накопления ключевых метаболитов помогает учёным одновременно профилировать обе стороны клеточного поведения: инструкции и вытекающую химию. Подход улучшает ранние методы выравнивания, упакован в доступный инструмент на Python и может быть расширен на другие пространственные технологии. По мере того как такие интегрированные карты станут более рутинными, они способны углубить наше понимание того, как опухоли и другие ткани организуют свой метаболизм, реагируют на лечение и эволюционируют во времени.
Цитирование: Xu, J., Shen, XT., Zhang, C. et al. haCCA: multi-module Integration of spot-based spatial transcriptomes and metabolomes. Commun Biol 9, 248 (2026). https://doi.org/10.1038/s42003-026-09526-w
Ключевые слова: пространственный мультиомикс, транскриптомика, метаболомика, метаболизм опухоли, интеграция данных