Clear Sky Science · ru
DPAS: оценка аномальности пептида, связанного с заболеванием, для выявления патогенных пептидов с помощью обучения одного класса
Почему крошечные фрагменты белков важны для нашего здоровья
Пептиды — короткие участки белков — становятся заметными игроками в современной медицине. Они могут выступать в роли точных передатчиков сигналов в организме и всё чаще используются как лекарственные средства и маркеры заболеваний. Однако определить, какие пептиды действительно связаны с болезнью, обычно можно лишь имея чёткие примеры как «больных», так и «здоровых» пептидов — чего биология редко предоставляет. В этом исследовании предложен новый способ выявления потенциально вредных пептидов, опирающийся только на уже известных пептидов, связанных с заболеванием, что даёт более быстрый и менее предвзятый путь к поиску будущих диагностических средств и терапий.
Проблема поиска группы «не связано с болезнью»
Традиционные компьютерные модели учатся, сравнивая две стороны: положительные примеры, известные как связанные с заболеванием, и отрицательные примеры, которые считаются безвредными. В исследованиях пептидов вторая группа вызывает трудности. Многие пептиды просто не были протестированы, поэтому маркировать их как «не связанные с болезнью» может вводить в заблуждение и вносить смещение. Предыдущие работы по противоопухолевым или противовоспалительным пептидам добивались впечатляющей точности, но часто опирались на вручную составленные или предположенные отрицательные наборы данных. В результате их модели могут плохо справляться с редкими сигналами или новыми типами пептидов, которые не похожи на данные обучения.
Учиться на том, что мы знаем, а не на догадках
Авторы идут другим путём: вместо того чтобы превращать задачу в двухстороннюю, они рассматривают пептиды, связанные с заболеванием, как единый связный набор и спрашивают: «Как этот набор выглядит в деталях?» Они собрали более 760 000 мутированных человеческих пептидов из специализированной базы данных, связанной с раком, и описали каждый пептид с помощью богатого набора признаков. Сюда входит частота каждого аминокислотного остатка, расположение пар аминокислот, базовые физико‑химические свойства, такие как объём и гидрофильность, а также короткие повторяющиеся последовательности, известные как мотивы. Метод главных компонент затем сжимает это многомерное описание в более управляемое представление, сохраняя основные источники вариативности.
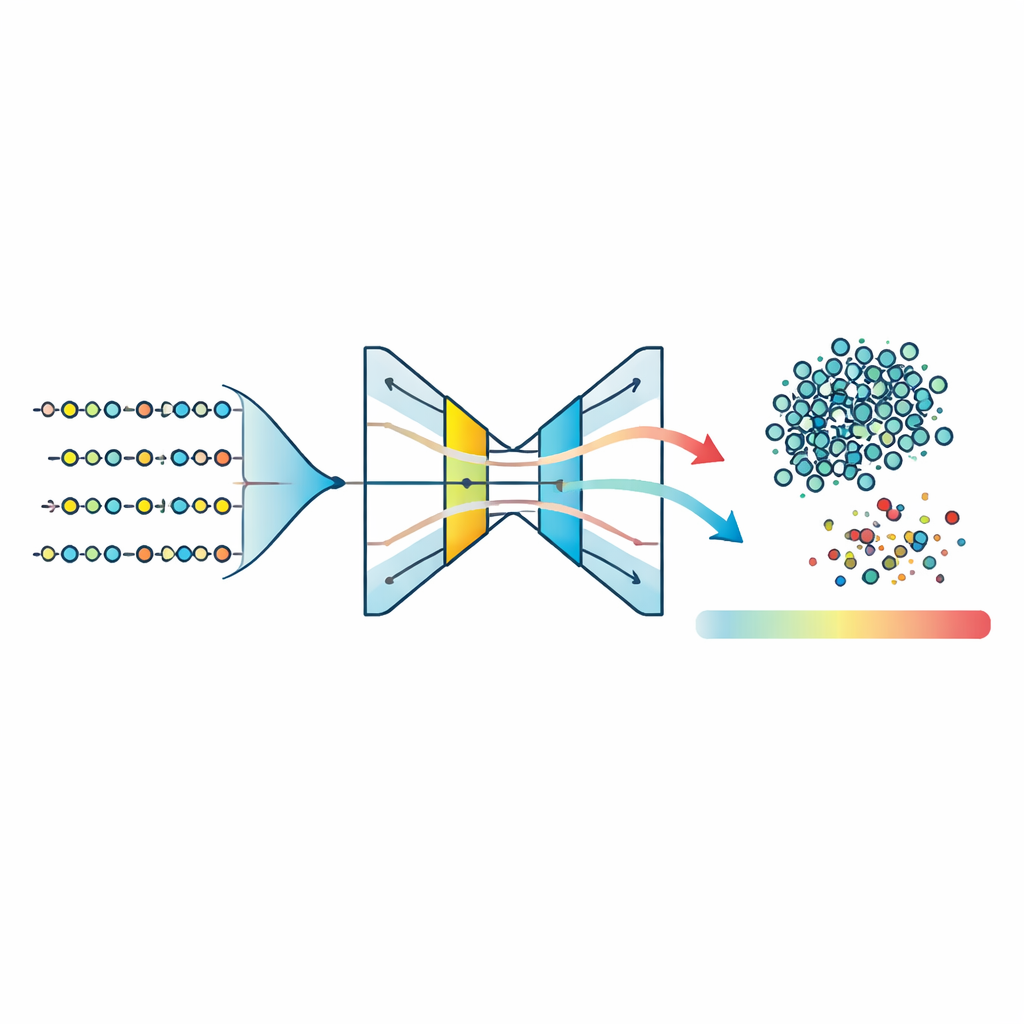
Выявление необычных пептидов моделями одного класса
Имея это сжатое пространство признаков, команда обучает три модели «одного класса» — алгоритма, предназначенные для изучения формы одного набора и отметки всего, что не вписывается в неё. Они тестируют одно-классовые опорные векторные машины, Isolation Forest и тип нейронной сети, называемый автоэнкодером. Автоэнкодер учится сжимать признаки каждого пептида до узкого внутреннего представления и затем восстанавливать их; пептиды, принадлежащие изученному заболеванию, восстанавливаются точно, в то время как необычные приводят к более высокой ошибке восстановления. Сравнение нормализованных скор аномалий по всем методам показывает, что автоэнкодер даёт наиболее плотный кластер типичных пептидов и самое чёткое разделение между инлайерами и аутлайерами. Установив порог по ошибке восстановления около 95-го процентиля, модель классифицирует большинство пептидов как вероятно связанных с заболеванием, одновременно стабильно помечая небольшую долю как нетипичные.
Преобразование сложных оценок в одно осмысленное число
Чтобы сделать результаты проще для биологической интерпретации, авторы вводят оценку аномальности пептида, связанную с заболеванием (DPAS). Эта оценка объединяет два компонента: насколько необычно пептид выглядит для автоэнкодера (его нормализованная ошибка восстановления) и насколько сильно его признаки влияют на предсказания, измеряемое популярным методом объяснимости SHAP. На практике особенно информативными оказываются мотивы и специфические физико‑химические свойства. DPAS комбинирует эти сигналы так, чтобы пептиды, которые одновременно структурно необычны и поддержаны биологически значимыми признаками, получали более высокие ранги. Пептиды с наивысшими оценками затем проверяются с помощью инструмента поиска мотивов, который связывает их с известными функциональными подписями, такими как сайты фосфорилирования, участки связывания металлов и другие регуляторные паттерны, часто вовлечённые в сигнальные и ферментативные процессы.
Что это значит для будущих диагностики и лекарств
Проще говоря, эта работа предлагает более умный фильтр для поиска подозрительных пептидов, не притворяясь, что мы знаем, какие из них определённо безвредны. Обучаясь только на подтверждённых примерах, связанных с заболеванием, и затем ранжируя новых кандидатов с помощью DPAS, исследователи могут приоритизировать короткий, биологически правдоподобный список пептидов для лабораторного тестирования. Многие из высокоранжированных кандидатов содержат хорошо известные функциональные мотивы, что усиливает идею об их возможной роли в болезненных процессах. Хотя метод всё ещё опирается на допущения и не имеет экспериментально подтверждённых «безопасных» пептидов для полной валидации, он обеспечивает более реалистичную и прозрачную основу для поиска пептидных биомаркеров и может быть адаптирован к другим типам биологических данных, где надёжных отрицательных примеров мало.
Цитирование: Khalid, Z., Khalid, R. & Sezerman, O.U. DPAS: disease-associated peptide anomaly score for identifying pathogenic peptides via one-class learning. Sci Rep 16, 9170 (2026). https://doi.org/10.1038/s41598-026-40099-0
Ключевые слова: пептиды, связанные с заболеваниями, обнаружение аномалий, автоэнкодер, поиск биомаркеров, обучение одного класса