Clear Sky Science · ru
Комплексные гетерозиготные мутации гена CHAT — миссенс и вариант в сплайс‑сайте — у двух братьев и сестры с врождённым миастеническим синдромом
Когда дыхание внезапно прекращается
Некоторым детям при рождении кажется здоровыми, но при небольшом повышении температуры они внезапно перестают дышать и требуют экстренной вентиляции. Для семей эти эпизоды оказываются пугающими и необъяснимыми. В этом исследовании проанализированы двое таких братьев и сестра из Японии: авторы проследили их жизнеугрожающие приступы слабости и апноэ (паузы дыхания) до мелких изменений в одном гене, который помогает нервам общаться с мышцами. Сочетая клинические данные, секвенирование генов и компьютерное моделирование структуры белка, исследователи показывают, как эти мутации, вероятно, нарушают ключевой фермент и дают врачам более чёткую цель для диагностики и лечения.
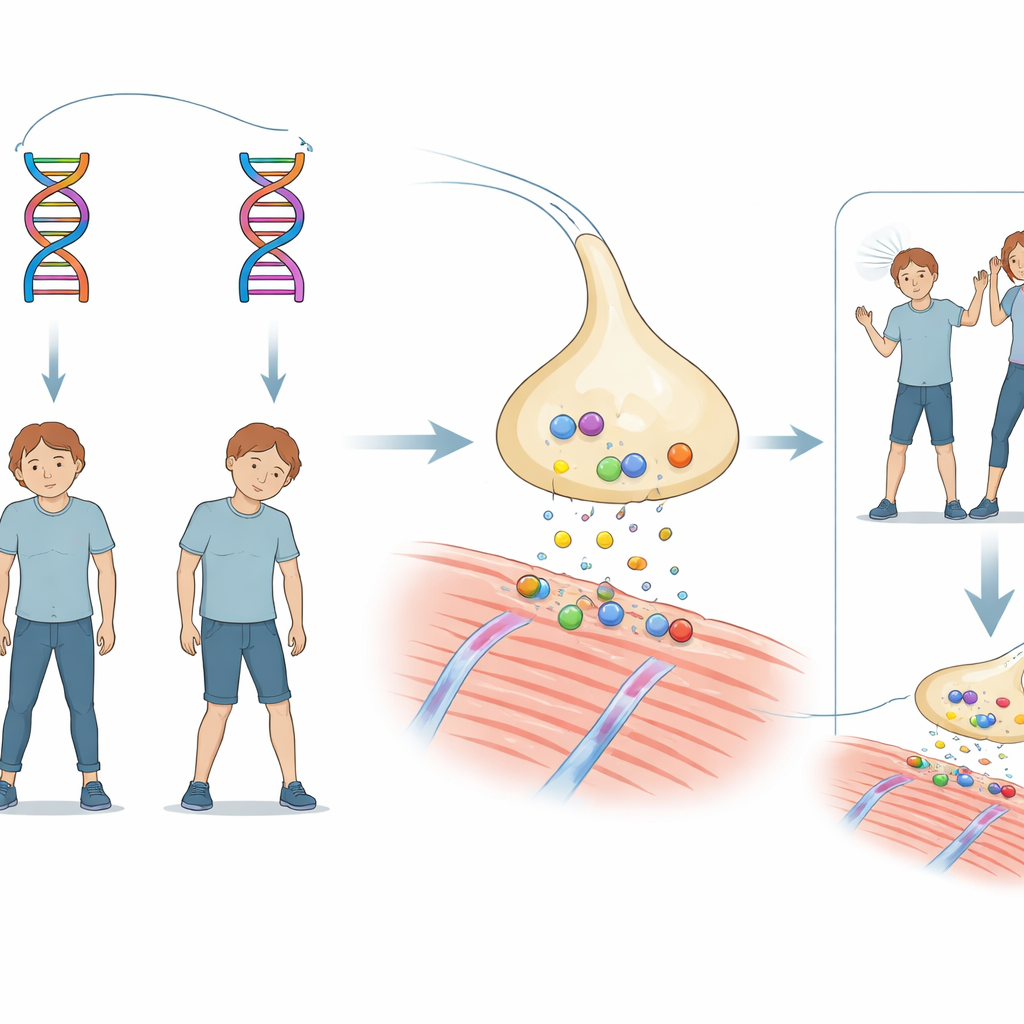
Семейная загадка внезапной слабости
История сосредоточена на брате и сестре, у которых в раннем детстве отмечалось несколько замедленное моторное развитие. Примерно в 18 месяцев у каждого возникали эпизоды апноэ и потери сознания при лихорадке, настолько серьёзные, что требовали подключения к аппарату искусственной вентиляции. По мере роста у обоих сохранялись приступы опущения век и генерализованной мышечной слабости, провоцируемые инфекциями, лихорадкой или физической нагрузкой. МРТ головного мозга была нормальной, и обычные антителозависимые формы миастении (заболевания, при котором нарушается связь между нервом и мышцей) были исключены. Тем не менее препарат, усиливающий химический сигнал между нервом и мышцей, явно улучшал их симптомы, что указывало на редкое наследственное состояние — врождённый миастенический синдром.
Поиск повреждённых инструкций
Чтобы выявить наследственную причину, команда секвенировала все белок-кодирующие гены у детей и их родителей. Они обнаружили, что каждый ребёнок нес по два разных изменения в одном и том же гене — CHAT, который кодирует холин ацетилтрансферазу — фермент, синтезирующий ацетилхолин, основной химический медиатор, используемый нервами для активации мышц. Одно изменение заменяло один строительный блок фермента (миссенс‑мутация, известная как G411R). Другое находилось в критической границе, где клетка обычно разрезает и склеивает участки гена при формировании РНК (мутация в сплайс‑сайте обозначена как c.752+2T>C). Каждый из родителей был носителем только одного из этих вариантов и не имел симптомов; только дети, унаследовавшие оба изменения, проявляли заболевание, что указывает на то, что пара мутаций совместно ослабляет функцию фермента.
Изучение того, как скрытый разрез меняет фермент
Поскольку исследователи не смогли получить достаточного количества естественной РНК CHAT из клеток крови, они использовали эксперимент с «минигeномом». Они клонировали соответствующий фрагмент гена в ДНК‑вектор, ввели нормальную или мутантную версию в культуру клеток и затем изучили, как обрабатывается РНК. В нормальном конструктазе РНК содержала все ожидаемые участки. В мутантной версии целый участок, известный как экзон 5, был пропущен, при этом рамка считывания гена оставалась неповреждённой. Это означало, что фермент будет синтезирован, но лишён короткого внутреннего фрагмента аминокислот. Сравнение с эволюционно родственными последовательностями показало, что этот отсутствующий участок сильно консервативен у разных видов, что указывает на его важную структурную роль.
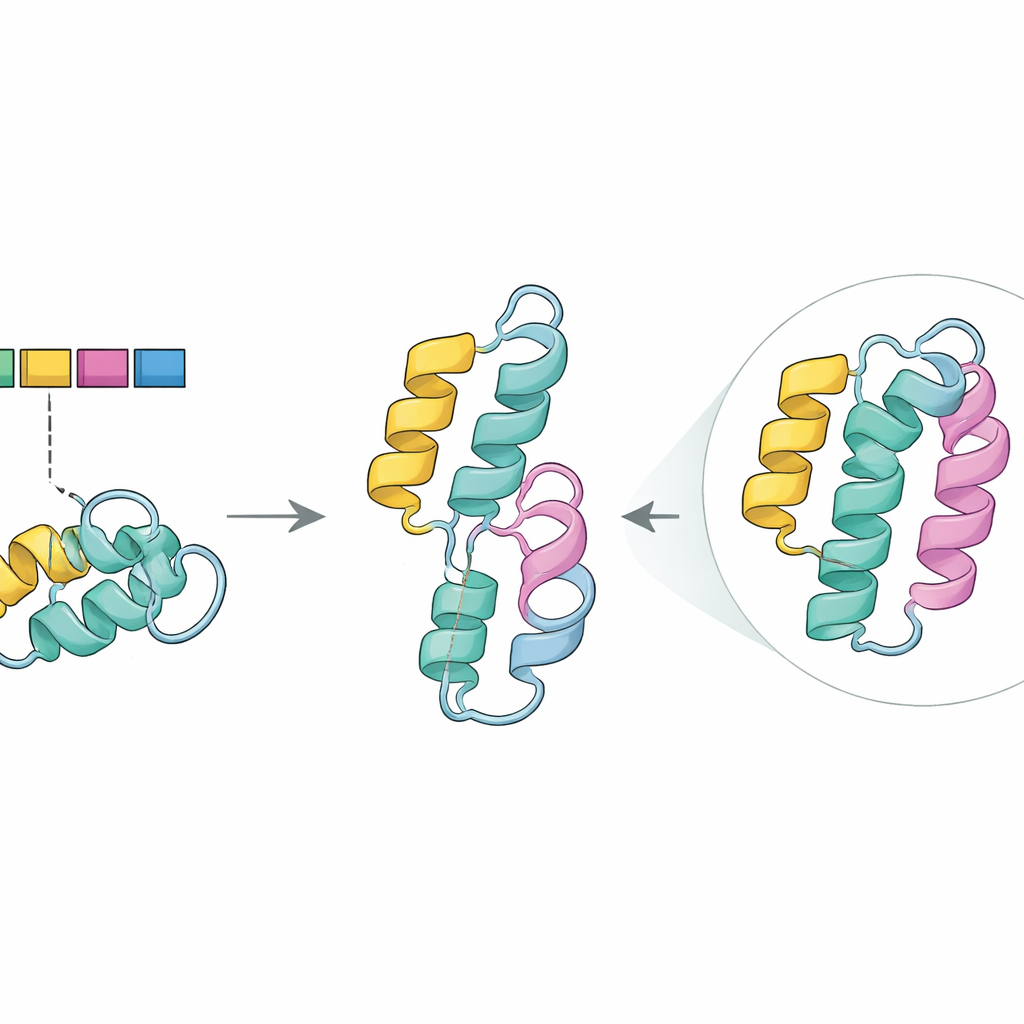
Наблюдая структурные повреждения in silico
Чтобы исследовать эту роль, команда обратилась к AlphaFold2 — продвинутой программе, предсказывающей трёхмерные структуры белков по их последовательностям. В нормальном ферменте участок, кодируемый экзоном 5, формирует одну из плотно упакованных спиральных структур (альфа‑спираль), которые помогают стабилизировать ядро белка. В предсказанной мутантной структуре эта спираль исчезла, оставив разрыв в области, ранее идентифицированной как ключевая для поддержания стабильности и обеспечения эффективного катализа. Вкупе с инструментами компьютерной оценки повреждающих мутаций эти результаты подтверждают идею, что пропуск экзона 5, особенно в сочетании с заменой G411R на другой копии гена, подрывает работу фермента, но не уничтожает её полностью — что согласуется с умеренно тяжёлым, но серьёзным клиническим проявлением у братьев и сестры.
Что это значит для пациентов и семей
Авторы приходят к выводу, что комбинация миссенс‑мутации G411R и недавно выявленного варианта в сплайс‑сайте гена CHAT весьма вероятно отвечает за врождённый миастенический синдром у этих детей. Демонстрируя с помощью минигенного анализа и структурного моделирования, как изменение в сплайс‑сайте удаляет стабилизирующую спираль из фермента, исследователи дают механистическое объяснение, на котором клиницисты и учёные могут опираться дальше. Для затронутых семей такое исследование значит больше, чем просто диагноз: оно обосновывает целенаправленное лечение препаратами, усиливающими нервно‑мышечную передачу, направляет генетическое консультирование при планировании последующих беременностей и добавляет важный пример в каталог того, как тонкие изменения в нашем генетическом коде могут существенно влиять на мышечную силу и базовый процесс дыхания.
Цитирование: Kikuchi, S., Wada, N., Mariya, T. et al. Compound heterozygous CHAT gene mutations, a missense and a splice site variant, in two siblings with congenital myasthenic syndrome. Sci Rep 16, 9346 (2026). https://doi.org/10.1038/s41598-026-39759-y
Ключевые слова: врождённый миастенический синдром, ген CHAT, холин ацетилтрансфераза, мутация в сплайс‑сайте, нервно‑мышечное синаптическое соединение