Clear Sky Science · ru
Математическое моделирование и вычисление NM‑многочленовых индексов для предсказания физико‑химических свойств
Почему это важно для будущих лекарств
Проектирование нового лекарства похоже на проектирование самолёта: хочется понять, как оно будет себя вести задолго до того, как его начнут изготавливать. Для препаратов это поведение включает летучесть, растворимость в воде или жирах и перемещение по организму. В этой статье показано, как продуманная математика может предсказывать многие из этих физических и химических свойств по структуре молекулы, что потенциально экономит время, деньги и сводит к минимуму методы проб и ошибок в поиске лекарств. 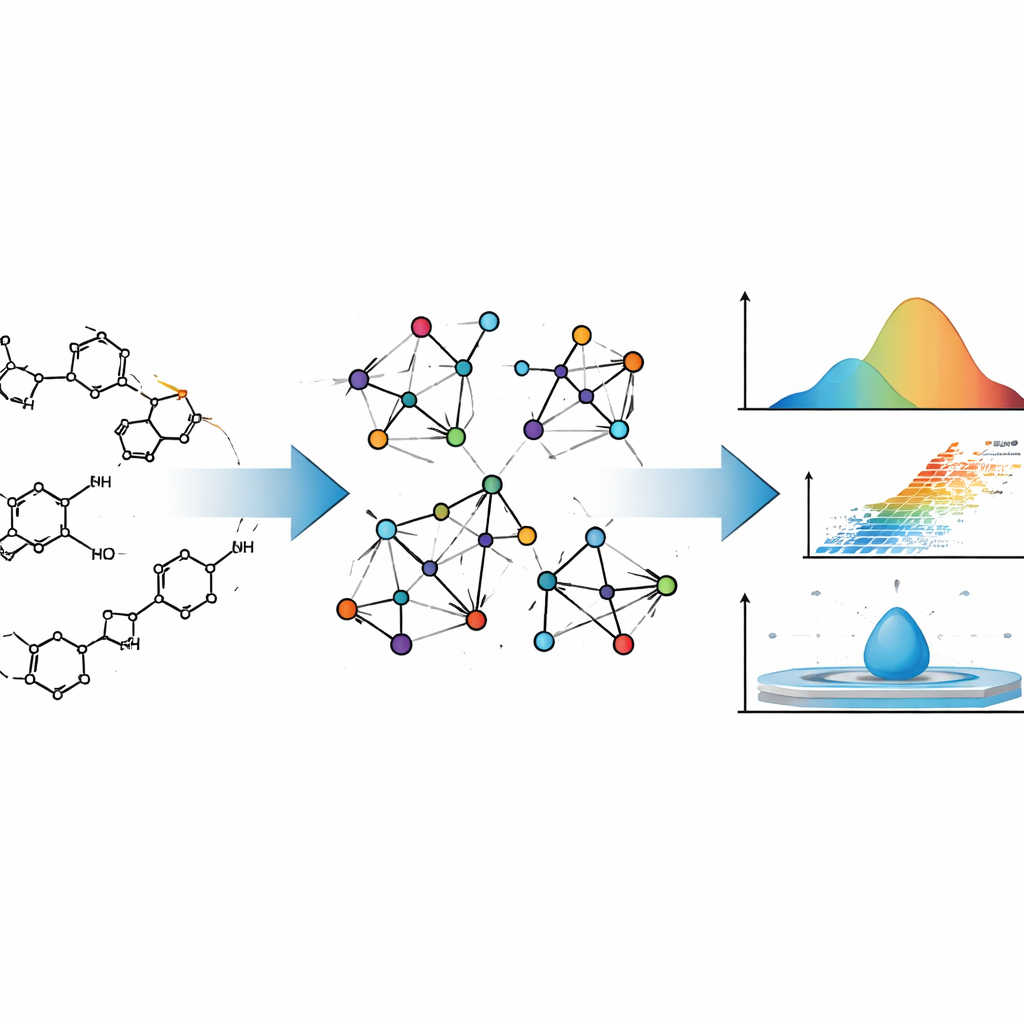
От молекул к сетям
Авторы рассматривают молекулы лекарств не просто как набор атомов, а как сети. В такой модели каждый атом — точка, а каждая химическая связь — линия, соединяющая две точки. Этот подход исходит из теории графов, раздела математики, изучающего сети всех видов — от связей в социальных сетях до энергосетей. Химики используют «молекулярные графы» десятилетиями, потому что некоторые числовые сводки этих графов — топологические индексы — часто коррелируют с поведением молекул в реальном мире, например с температурой кипения или плотностью.
Добавление информации о соседях
Традиционные индексы обычно учитывают только число связей у каждого атома. Команда авторов пошла дальше. Они используют так называемые NM‑многочленовые (neighborhood M‑polynomial) индексы, которые описывают не только собственные связи атома, но и то, насколько связаны его соседи. Это более богатое представление фиксирует тонкие особенности, например степень разветвлённости молекулы, слияние колец и расположение атомов кислорода или азота в каркасе. Эти элементы в свою очередь влияют на то, как сильно молекулы притягиваются друг к другу, насколько они жёсткие и как их электроны реагируют на электрические поля — все это определяет ключевые физико‑химические свойства.
Проверка идеи на реальных противораковых препаратах
Чтобы связать математику с реальными данными, авторы сначала вычисляют NM‑многочленовые индексы для двух хорошо известных противораковых препаратов — митоксантрона и доксорубицина. Оба представляют собой сложные полициклические структуры, широко применяемые в химиотерапии. Переведя детальные химические формулы в молекулярные графы, а затем в NM‑многочленовые индексы, авторы демонстрируют, как метод систематически отслеживает структурные изменения при разных «размерах» молекул. Затем процесс автоматизируют с помощью программы на Python: по матрице смежности молекулы она мгновенно возвращает полный набор индексов, минимизируя человеческие ошибки и ускоряя расчёты, которые вручную были бы утомительны. 
Обучение машин чтению «молекулярных отпечатков»
Далее исследователи расширяют набор данных до 45 полициклических лекарственных средств, включая знакомые названия — ацетаминофен, ибупрофен и несколько современных таргетных препаратов. Для каждой молекулы они вычисляют девять NM‑многочленовых индексов и собирают девять экспериментально измеренных свойств: сложность, температуру кипения, энтальпию испарения, точку вспышки, молярную ретрактность, поляризуемость, поверхностное натяжение, молярный объём и показатель преломления. Затем они обучают несколько регрессионных моделей в стиле машинного обучения — линейную, Ridge, Lasso и Elastic Net — чтобы сопоставить комбинации индексов с каждым свойством. На всём протяжении используются строгие статистические меры: удаление избыточных входных переменных, стандартизация, повторная кросс‑валидация на 80 % данных и тестирование финальных моделей на не затронутых 20 %.
Что показывают числа
Модели демонстрируют, что NM‑многочленовые индексы особенно эффективны для свойств, связанных с упаковкой и взаимодействием молекул. Для температуры кипения, энтальпии испарения, точки вспышки, молярной ретрактности, поляризуемости и молярного объёма лучшие модели достигают очень высокой корреляции с экспериментальными значениями. Регуляризованные методы, такие как Ridge и Elastic Net, в целом показывают лучшие результаты, что указывает на пользу лёгкого ограничения модели для выделения наиболее информативных аспектов индексов. Тепловая карта корреляций подтверждает, что несколько индексов — особенно те, что связаны с общей связностью и «богатостью соседей» — стабильно сильно коррелируют с этими свойствами в панели из 45 препаратов.
Ограничения и пути улучшения
Не все свойства поддаются хорошему предсказанию. Показатель преломления, характеризующий, как свет отклоняется при входе в вещество, оказался упорным: модели плохо превосходят простые средние, и NM‑многочленовые индексы с ним слабо коррелируют. Поверхностное натяжение описано умеренно, но не так чётко, как другие свойства. Эти пробелы указывают на то, что части поведения зависят от факторов, выходящих за рамки двумерной связности — например трёхмерной геометрии или тонких электронных эффектов. Авторы предлагают в будущих работах сочетать NM‑многочленовые индексы с квантово‑химическими или 3D‑дескрипторами, чтобы преодолеть это ограничение.
Что это значит для дизайна лекарств
Проще говоря, исследование показывает, что сложная, но структурированная математика способна превратить статический набросок молекулы в внезапно точный предсказатель её поведения в лаборатории. Для ряда важных свойств — температуры кипения, объёма молекулы или лёгкости смещения электронов — подход с NM‑многочленами в сочетании с современными регрессионными методами сопоставим или превосходит более ранние методы с простыми индексами или малыми наборами данных. Хотя это ещё не полная замена экспериментам, метод даёт конструкторам лекарств быстрый инструмент скрининга: вычисляя такие графовые «отпечатки», можно заранее оценивать ключевые физико‑химические свойства, сосредоточить лабораторную работу на перспективных кандидатах и эффективнее исследовать химическое пространство.
Цитирование: Tawhari, Q.M., Naeem, M., Koam, A.N.A. et al. Mathematical Modeling and Computation of NM-Polynomial Indices for Physicochemical Properties Prediction. Sci Rep 16, 8136 (2026). https://doi.org/10.1038/s41598-026-39562-9
Ключевые слова: теория химических графов, прогнозирование свойств лекарств, молекулярная топология, машинное обучение в химии, физико‑химические дескрипторы