Clear Sky Science · ru
CiCLoDS: Совместная кластеризация клеток и отбор генов для пространственной транскриптомики одиночных клеток
Поиск «районов» в городе клеток
Современные микроскопы теперь могут определять, какие гены активны в сотнях тысяч клеток, сохраняя при этом их исходное положение в ткани. Эта революция в пространственной транскриптомике похожа на превращение размытой карты города в уличный вид каждого дома. Но есть загвоздка: такие карты содержат измерения тысяч генов на клетку — намного больше, чем учёным удобно интерпретировать или чем они могут позволить себе измерять в последующих экспериментах. В этом исследовании представлен CiCLoDS — новый метод, который находит значимые клеточные «районы» и одновременно выбирает небольшой, интерпретируемый набор генов, определяющих эти районы.
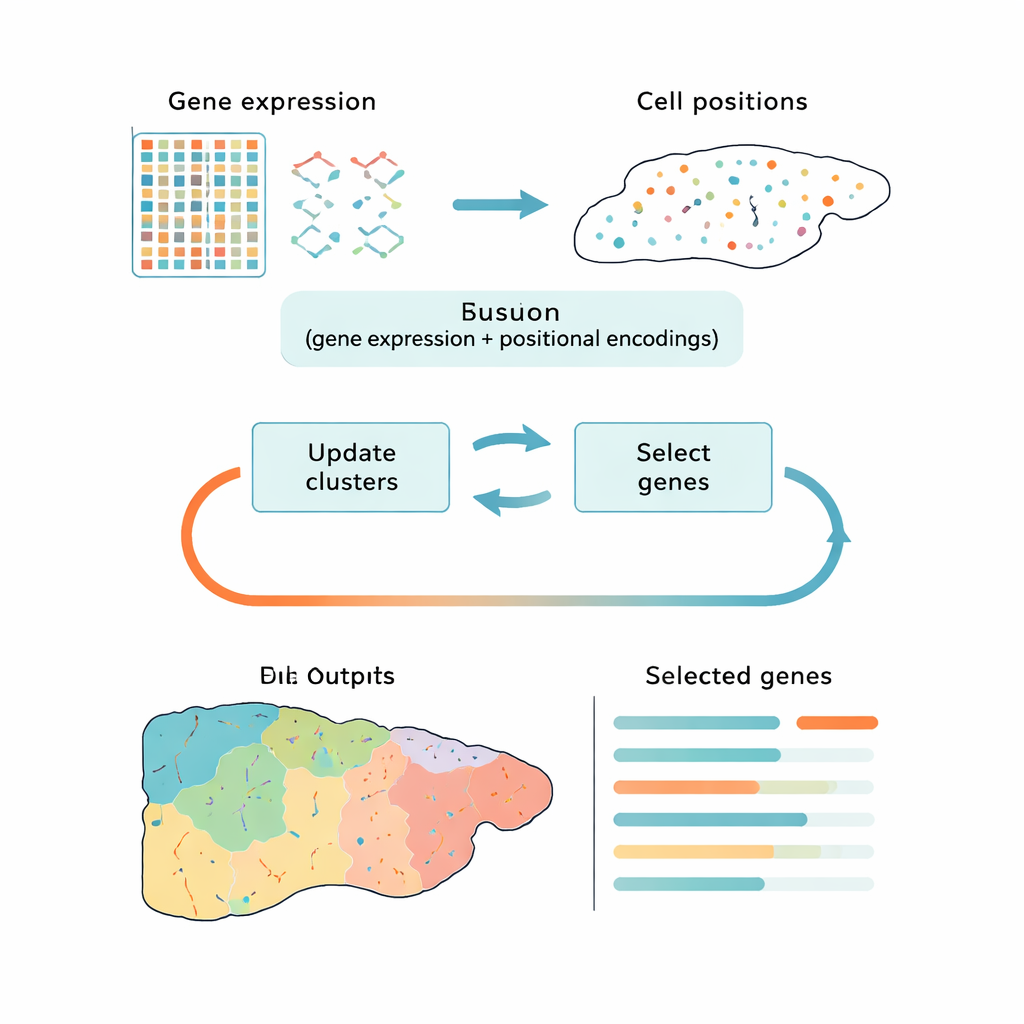
Более умный способ сокращать большие данные
Большинство современных инструментов решают эту задачу в два несвязанных шага: сначала упрощают данные, затем группируют клетки в кластеры. Популярные подходы, такие как метод главных компонент (PCA), сохраняют общую вариацию, но могут фокусироваться на техническом шуме или общих сигналах клеточного цикла, а не на биологических различиях, которые имеют значение. Другие методы используют глубокое обучение для поиска закономерностей, но действуют как «чёрные ящики» и не показывают явно, какие гены наиболее важны. CiCLoDS идёт иным путём: он рассматривает отбор генов и кластеризацию как единую задачу в рамках пользовательского «бюджета» на число оставляемых генов. По сути, метод отвечает на вопрос: какой ограниченный набор генов лучше всего объясняет, как клетки распределяются по группам, учитывая их активность генов и, при наличии, их физическое положение в ткани?
От математики к картам реальных тканей
Авторы адаптировали семейство математически прозрачных методов, называемых субпространственной кластеризацией, к реалиям пространственной транскриптомики, где наборы данных могут насчитывать более миллиона клеток. CiCLoDS работает с простой таблицей «клетка‑по‑гену», назначая клетки кластерам и оценивая вклад каждого гена в разделение этих кластеров. Метод также может учитывать пространственную информацию, добавляя позиционные «кодировки», описывающие, где находится каждая клетка в ткани, не меняя при этом основного алгоритма оптимизации. На больших наборах данных мышиной печени и человеческого толстой кишки, полученных с помощью высокоразрешающих платформ визуализации, CiCLoDS выполняется за считанные минуты на стандартных компьютерах и даёт компактные генетические панели — порядка нескольких десятков до сотен генов — которые при этом сохраняют богатую структуру исходных данных.
Выявление скрытых зон и кровеносных сосудов
Применив CiCLoDS к мышиной печени, авторы проверяли, может ли метод восстановить известные «зонационные» шаблоны — постепенные изменения функций гепатоцитов от одной стороны лобуля к другой. По сравнению с PCA и ведущим инструментом отбора генов geneBasis, CiCLoDS дал более чёткие пространственные зоны с резкими границами и значительно меньшим числом неправильно отнесённых областей, что подтверждается количественными метриками согласования с эталонной картой. Замечательно, что при увеличении числа разрешённых генов CiCLoDS заново выявил группы гепатоцитов, похожие на пери-портальные и пери-центральные, которые тесно соответствовали экспертно определённым референсным кластерам, даже несмотря на то, что метод не знал о ключевом маркёрном гене AXIN2 и не получал явных пространственных координат. При добавлении пространственных кодировок CiCLoDS также отобрал наборы генов, обогащённые функциями, связанными с поверхностью клеток и сосудами, и смог точно отличать реальные кровеносные сосуды от артефактов визуализации — то, с чем более простые методы либо не справились, либо добивались только при помощи дополнительных эвристик.
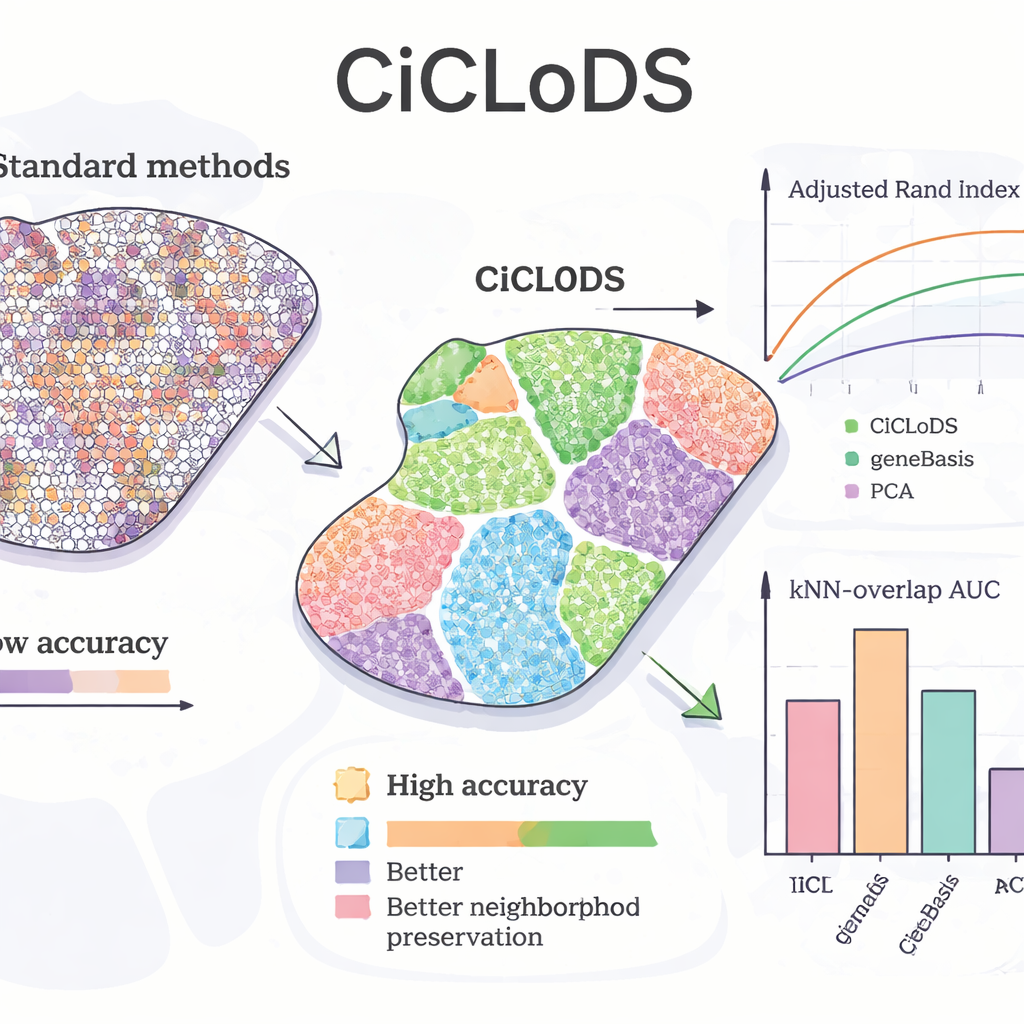
Обобщение на разные ткани и улучшение других методов
Чтобы проверить, сохраняется ли эффективность CiCLoDS на сильно отличающихся тканях и у разных доноров, авторы проанализировали образцы дорсолатеральной префронтальной коры человека от трёх доноров. Здесь CiCLoDS показал результаты не хуже, а порой лучше, чем специализированные пространственные методы, такие как BayesCafe и BayesSpace, особенно на сложном образце, где другие инструменты испытывали трудности. В исследовании также подчёркивается «гибридный» подход: сначала запуск CiCLoDS для получения устойчивых кластеров, а затем использование этих кластеров в BayesSpace. Эта стратегия «тёплого старта» повысила общую точность и дала слоистые паттерны мозга, которые лучше всего соответствовали экспертным аннотациям, показывая, что CiCLoDS может как работать автономно, так и делать дальнейшие вероятностные модели более надёжными.
Почему это важно для биологии и медицины
Для неспециалистов ключевой вывод в том, что CiCLoDS превращает перегруженные клеточные карты в сжатые, биологически информативные сводки. Вместо работы с тысячами шумных измерений исследователи получают управляемый список генов и чёткие пространственные кластеры, отражающие реальную организацию ткани — метаболические зоны в печени, кровеносные сосуды и их ниши, а также слоистые структуры в мозге. Поскольку «бюджет» на гены контролируется пользователем, а вычисления лёгки, CiCLoDS может помочь в проектировании целевых панелей генов для будущих экспериментов, облегчить интерпретацию сложных пространственных наборов данных и обеспечить надёжные отправные точки для более сложного моделирования. В эпоху, когда узким местом становится не сбор данных, а их понимание, такие инструменты, как CiCLoDS, обещают сделать высокоразмерные карты тканей и практичными, и информативными.
Цитирование: Wang, N., He, Y., Ray, E. et al. CiCLoDS: Joint cell clustering and gene selection for single-cell spatial transcriptomics. Sci Rep 16, 5356 (2026). https://doi.org/10.1038/s41598-026-39168-1
Ключевые слова: пространственная транскриптомика, кластеризация клеток, выбор панелей генов, архитектура тканей, анализ одиночных клеток