Clear Sky Science · ru
Молекулярная характеристика рецессивно наследуемых атактических и невропатических расстройств в родственниках по крови из Пакистана
Почему это важно для семей и врачей
Проблемы с равновесием, ходьбой и чувствительностью в руках и ногах могут серьёзно ограничивать жизнь, особенно когда начинаются в детстве и постепенно прогрессируют в течение многих лет. Для многих семей, особенно в регионах, где распространены браки между родственниками, такие симптомы передаются из поколения в поколение без ясного объяснения. Это исследование отвечает на насущный вопрос для таких семей в Пакистане: может ли современный анализ ДНК наконец выявить скрытые генетические причины их атаксии (нарушения равновесия и координации) и периферической невропатии (повреждения нервов конечностей) и помочь врачам поставить точный диагноз и предложить возможные варианты лечения?
Прослеживание наследственной болезни в больших семьях
Исследователи работали с семью большими пакистанскими семьями, в которых несколько членов страдали выраженными двигательными и нервными нарушениями. У одних преобладала атаксия, что затрудняло устойчивую ходьбу или управление речью и движениями глаз. У других были классические признаки периферической невропатии: атрофия мышц рук и ног, деформации стоп и утрата рефлексов. В этих семьях родители были родственниками, что повышало вероятность унаследования детьми двух копий одной и той же редкой дефектной версии гена. На основе образцов крови от больных и здоровых родственников команда провела секвенирование экзома — чтение почти всех кодирующих белки участков генома — чтобы найти вредоносные изменения, которые совпадали бы с картой болезни в родословных. 
Выявление редких патогенных генов
Отфильтровав частые и безвредные варианты ДНК, учёные обнаружили вероятные причинно‑значимые варианты у пяти из семи семей. В каждой из этих семей присутствовало собственное специфическое генетическое изменение, и все они соответствовали рецессивному наследованию: заболевание развивалось только при наличии двух дефектных копий — по одной от каждого родителя. В одной семье с развитием нарушений равновесия и речи у взрослых причиной оказался редкий вариант в гене MFSD8, который участвует в транспортировке материалов в клетки утилизации — лизосомы. В другой семье вредоносный вариант в AFG3L2, белке, отвечающем за поддержание здоровья митохондрий — «энергетических станций» клетки, — был связан с детским началом спастической атаксии с дистонией (патологическими мышечными сокращениями). Третья семья несла сдвиг рамки считывания в SETX — гене, защищающем ДНК во время репарации, уже известном как причина атаксии с окуломоторной апраксией, расстройства, поражающего также движения глаз.
Более детальный взгляд на наследственное повреждение нервов
Ещё в двух семьях обнаружилась форма болезни Шарко‑Мари‑Тута (CMT) — группы наследственных заболеваний, которые повреждают длинные нервы, иннервирующие стопы и кисти. В обеих семьях исследователи нашли патогенные варианты в гене GDAP1, важном для нормальной митохондриальной функции в нервных клетках. Один вариант в GDAP1 приводил к укорочению белка и ассоциировался с очень тяжёлым ранним началом заболевания; другой — замена одного аминокислотного звена — давал несколько более мягкое течение. Примечательно, что самый тяжело поражённый пациент в одной из семей с CMT также был гомозиготен по известному патогенному варианту во втором гене, MMACHC, который участвует в метаболизме витамина B12 и иногда поддаётся лечению витаминными препаратами. Такое сочетание вариантов может объяснить, почему его симптомы были тяжелее, чем у родственников без варианта MMACHC.
Когда поиск по ДНК не даёт ответа
Не во всех семьях удалось получить однозначный генетический ответ. В двух из семи семей команда не нашла ни одного изменения в экзоме, которое убедительно соответствовало бы картине наследования болезни. В одном случае был обнаружен вариант в гене EHHADH, который совпадал с паттерном наследования, но прогнозировался как безвредный и известен как причина другого заболевания, связанного с почками, при нарушении его функции. В другом случае два кузена с похожими двигательными нарушениями оказались вызваны разными причинами: один мальчик нес известный патогенный вариант в ALS2, который может приводить к ювенильным формам болезни мотонейронов, в то время как его поражённые кузены этого варианта не имели. Эти неразрешённые случаи указывают на то, что важные мутации могут находиться в регионах генома, не покрываемых стандартным секвенированием экзома, либо что несколько тонких генетических факторов могут взаимодействовать между собой.
Что это означает для пациентов и будущей помощи
В сумме результаты показывают, что мощные методы анализа ДНК способны раскрыть конкретные гены, лежащие в основе сложных нарушений нервной системы и равновесия, даже в условиях с ограниченными ресурсами. Для пяти семей с чёткими результатами работа превращает расплывчатые ярлыки вроде «атаксия» или «невропатия» в точные диагнозы, связанные с конкретными генами, что помогает генетическому консультированию, уточнению прогноза и в некоторых случаях указывает на варианты лечения, такие как терапия, связанная с витамином B12 при заболеваниях, связанных с MMACHC. Исследование также расширяет понимание учёных о том, как гены MFSD8, AFG3L2, SETX, GDAP1, MMACHC и ALS2 влияют на здоровье нервных клеток головного и спинного мозга, а также периферических нервов. В перспективе потребуется более полное секвенирование генома и функциональные исследования, чтобы разгадать оставшиеся загадки и превратить эти генетические открытия в более раннюю диагностику и лучшее лечение для поражённых детей и взрослых. 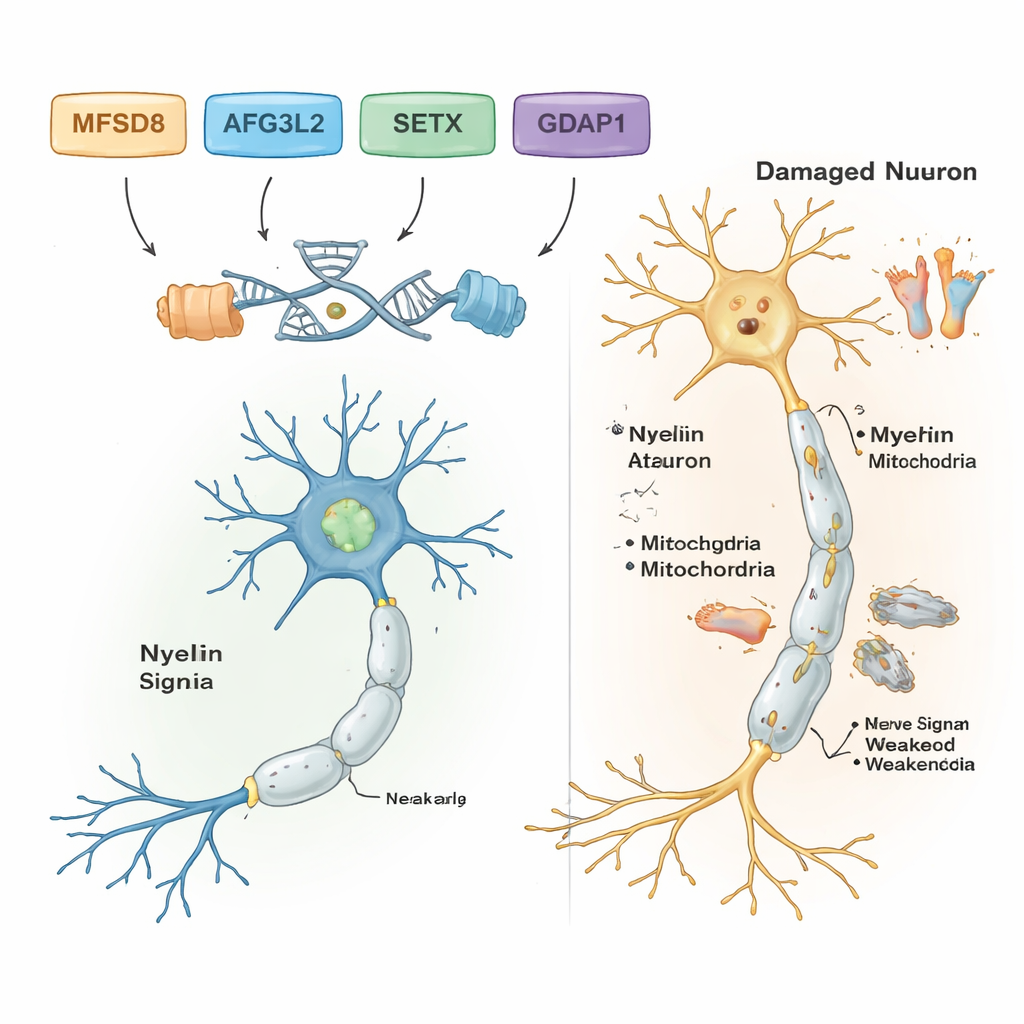
Цитирование: Aslam, F., Wajid, M., Butt, A.I. et al. Molecular characterization of recessively inherited ataxic and neuropathic disorders in consanguineous Pakistani families. Sci Rep 16, 6529 (2026). https://doi.org/10.1038/s41598-026-37808-0
Ключевые слова: атаксия, периферическая невропатия, секвенирование экзома, болезнь Шарко — Мари — Тута, генетическая диагностика