Clear Sky Science · ru
Оптимизация отбора признаков в данных микрочипов рака с помощью управляемого кучей эволюционного фреймворка для высокоразмерных пространств
Почему важно правильно выбирать гены
Тесты на рак, основанные на современных генетических технологиях, могут измерять десятки тысяч генов одновременно, тогда как у врачей часто есть данные только от нескольких десятков пациентов. В этой огромной «генной чаще» скрывается гораздо меньшее число сигналов, которые на самом деле отделяют один вид рака от другого или опухоль от здоровой ткани. В статье предлагается новый метод умного поиска для автоматического выделения этих ключевых генов с целью сделать компьютерную помощь в диагностике рака более точной, быстрой и удобной для интерпретации.
Слишком много сигналов и слишком мало данных
Эксперименты на микрочипах и похожие технологии позволяют исследователям измерять уровни активности тысяч генов в каждой пробе пациента. Тем не менее количество образцов обычно очень невелико, порой меньше ста. Многие показания по генам шумны, избыточны или не имеют отношения к рассматриваемому заболеванию. Сохранение всех признаков может перегрузить алгоритмы обучения, замедлить вычисления и привести к вводящим в заблуждение моделям, которые цепляются за случайные случайности вместо истинной биологии. Процесс сокращения набора до полезного подмножества называется «отбор признаков», и он критически важен для получения надежных предсказаний из высокоразмерных медицинских данных.
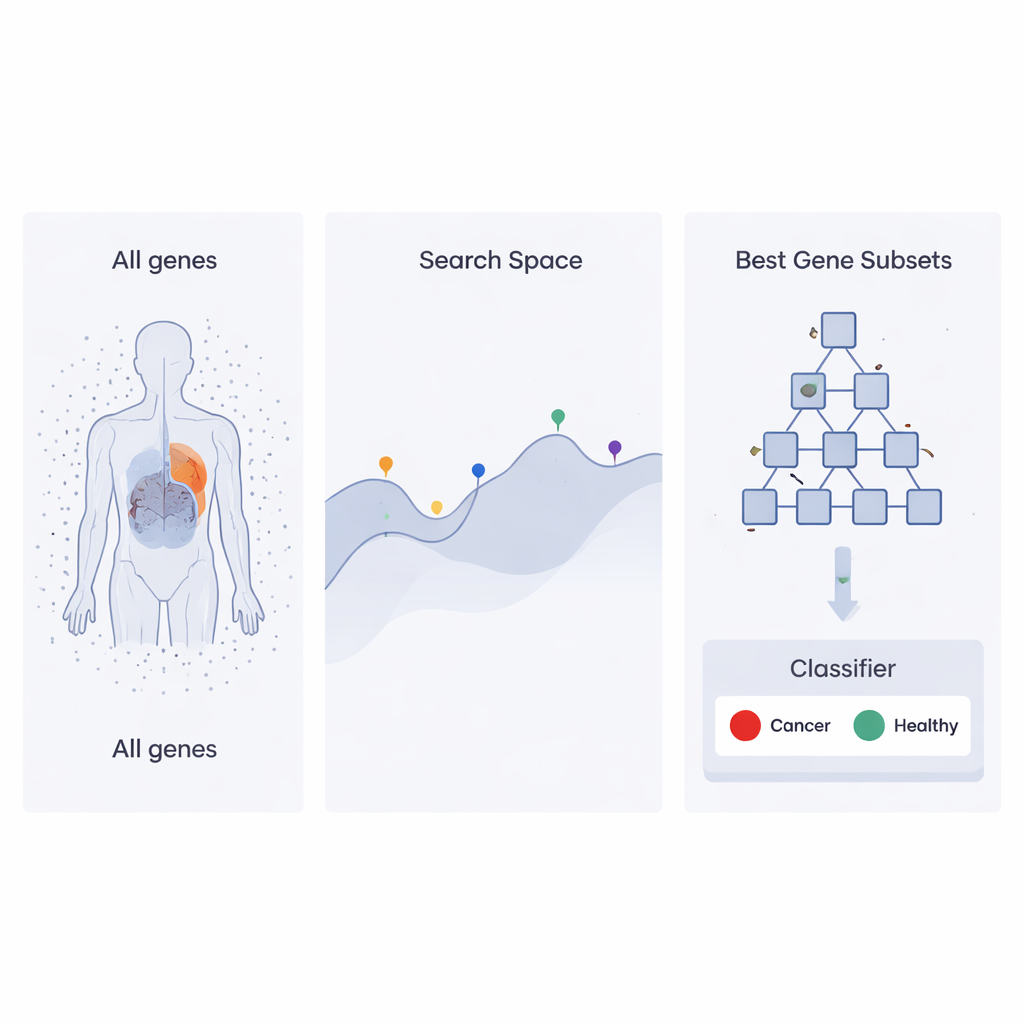
Стратегия поиска, вдохновлённая корпоративной иерархией
Авторы опираются на недавний подход оптимизации, называемый Heap‑Based Optimizer (HBO), который заимствует идеи из того, как организованы сотрудники в компании. Представьте каждое возможное множество генов как «сотрудника», чья эффективность оценивается по тому, насколько хорошо оно помогает классификатору отличать образцы рака от здоровых. Эти «сотрудники» располагаются в иерархии, подобной служебной лестнице, с использованием компьютерной структуры, известной как куча. Наборы генов с высокой эффективностью занимают верхние позиции, а более слабые — нижние. В ходе многих итераций низко расположенные «сотрудники» подстраивают свои варианты, копируя и слегка модифицируя то, что делают их начальники и коллеги, постепенно двигая всю «организацию» к лучшим решениям.
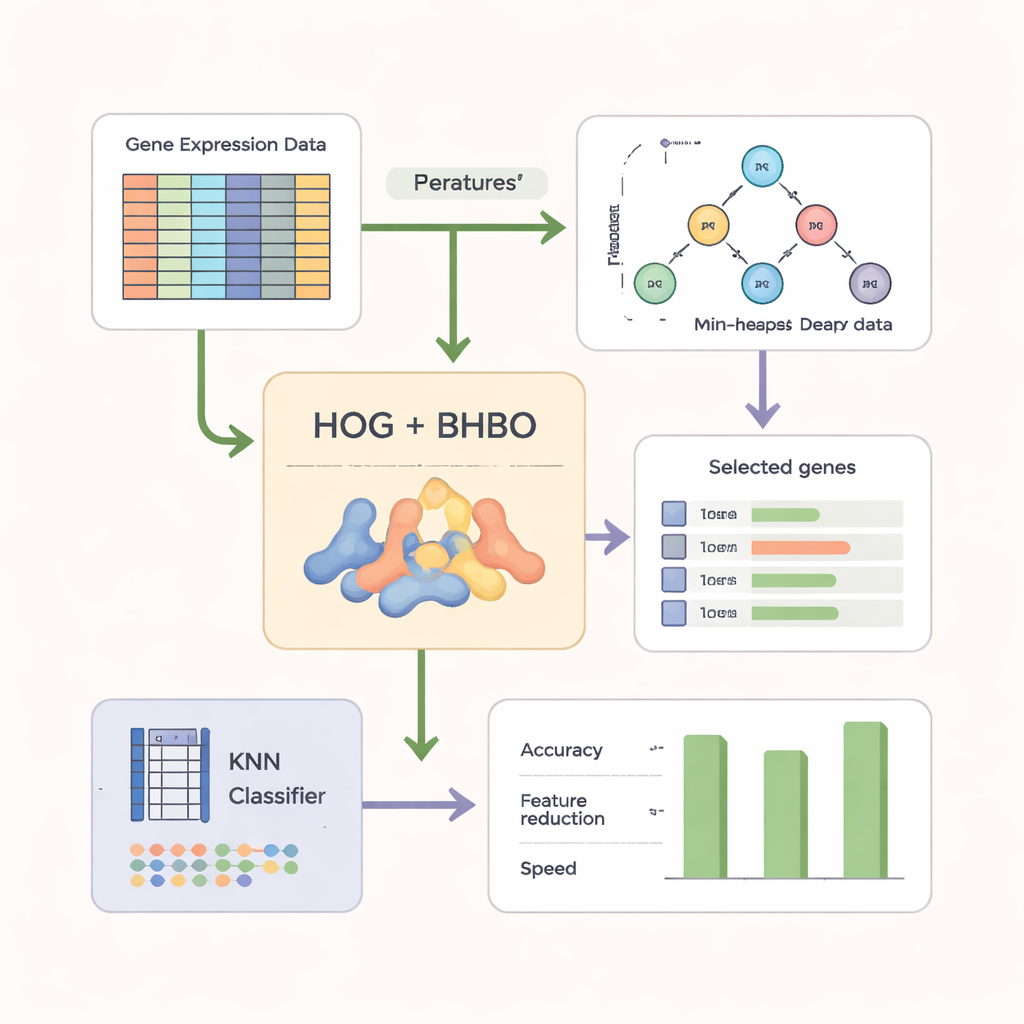
Преобразование сырых генетических данных в более чёткие шаблоны
Чтобы сделать поиск эффективнее, авторы не полагаются исключительно на сырые показания генов. Они сначала преобразуют данные микрочипов в форму, похожую на изображение, и применяют технику Histogram of Oriented Gradients (HOG), широко используемую в компьютерном зрении. HOG фиксирует, как меняются уровни экспрессии по генам, подчёркивая локальные паттерны вместо изолированных измерений. Эти признаки на основе шаблонов затем комбинируются с исходной информацией о генах. В качестве «судьи» используется простой классификатор k‑Nearest Neighbors (KNN), оценивающий каждое кандидатное подмножество генов по точности классификации новых образцов, а также поощряющий более компактные наборы.
Тестирование на нескольких наборах данных о раке
Исследователи оценили бинарную версию Heap‑Based Optimizer (BHBO) на девяти общедоступных наборах микрочиповых данных по раку, включая опухоли мозга, лейкемии, рак простаты и смешанные коллекции опухолей с множеством подтипов. Каждый набор содержал от тысяч до более пятнадцати тысяч измеренных генов, но относительно небольшое число образцов пациентов. Для каждого набора BHBO запускали многократно и сравнивали с семью известными методами поиска, такими как генетические алгоритмы и рой частиц. Команда измеряла не только точность, но и количество оставленных генов, скорость сходимости поиска и устойчивость результатов при моделируемых искажениях данных — шуме, пакетных эффектах и ошибках в метках.
Чего добился новый метод
По результатам на девяти наборах, подход, управляемый кучей, достиг в среднем примерно 95-процентной точности классификации, одновременно сокращая число генов более чем на 85 процентов. Он явно превосходил конкурентов на ряде наборов и демонстрировал более быструю сходимость, то есть находил хорошие наборы генов за меньшее число шагов поиска. Даже когда авторы намеренно портировали данные — добавляя шум или меняя метки некоторых образцов — производительность метода падала лишь незначительно и оставалась лучше альтернатив. Статистические тесты подтвердили, что эти улучшения вряд ли являются случайностью.
Что это значит для будущей диагностики рака
В практическом плане работа показывает, что грамотно спроектированная стратегия поиска способна просеять огромные генетические наборы данных и выявить небольшие информационно насыщенные панели генов, которые по‑прежнему хорошо классифицируют рак. Для клиницистов и исследователей такие компактные наборы генов проще биологически верифицировать, дешевле измерять в последующих тестах и удобнее интегрировать в инструменты поддержки принятия решений. Хотя метод не открывает непосредственно новые лекарства или пути, он усиливает фокус на многообещающих генетических маркерах, помогая последующим исследованиям сосредоточиться на наиболее информативных сигналах, скрытых в высокоразмерных данных о раке.
Цитирование: Alweshah, M., Jebril, H., Kassaymeh, S. et al. Optimizing feature selection in cancer microarray data using a heap-driven evolutionary framework for high-dimensional spaces. Sci Rep 16, 6726 (2026). https://doi.org/10.1038/s41598-026-37803-5
Ключевые слова: микрочипы рака, отбор признаков, метаэвристическая оптимизация, генные биомаркеры, интеллектуальный анализ медицинских данных