Clear Sky Science · ru
Гибридная глубокая обучающая архитектура для точной классификации высокоразмерных геномных данных
Осмысление лавины геномных данных
Современные методы анализа ДНК позволяют измерять десятки тысяч генов в одном эксперименте, что обещает более раннюю диагностику заболеваний и более точные методы лечения. Однако такой объём данных настолько велик, шумен и сложен, что даже мощные вычислительные модели часто затрудняются найти ясные и надёжные закономерности. В этой статье предлагается новый тип системы искусственного интеллекта, специально разработанный для работы с такими непростыми геномными данными: цель — повысить точность прогнозов и одновременно объяснить, как эти прогнозы получены.
Почему геномные данные трудно использовать
Геномные исследования регулярно дают гораздо больше измерений, чем пациентов или образцов. Многие из этих измерений неважны, дублируют друг друга или искажены техническим шумом. Традиционные методы машинного обучения либо требуют, чтобы эксперты вручную выбирали, какие гены могут иметь значение, либо пытаются использовать всё подряд и рискуют переобучиться — хорошо работать на тренировочных данных, но давать плохие результаты на новых случаях. Глубокое обучение, преобразившее такие области, как распознавание изображений, умеет автоматически выделять закономерности из сырых данных. Однако в геномике оно часто ведёт себя как «чёрный ящик»: может давать точные ответы, но даёт мало понимания причин, что ограничивает его принятие в медицине, где прозрачность имеет решающее значение.
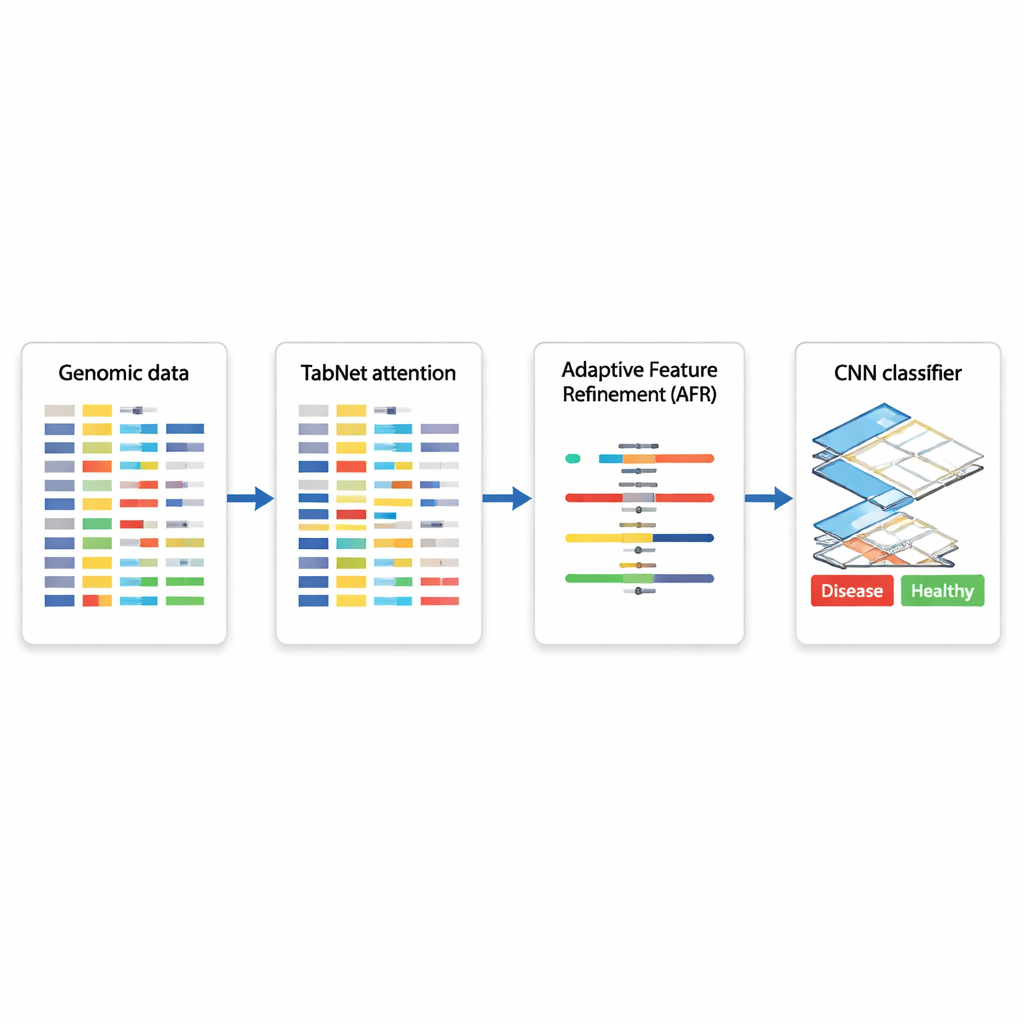
Гибридная архитектура ИИ для генетических решений
Авторы предлагают гибридную глубокую архитектуру, объединяющую три специализированных модуля. Сначала компонент под названием TabNet действует как прожектор, просматривая все доступные геномные измерения и выявляя, какие признаки наиболее информативны для конкретной задачи — например, для различения раковой и нераковой ткани. Вместо равного обращения со всеми генами, TabNet фокусируется на разреженном подмножестве, которое кажется наиболее релевантным. Затем слой Adaptive Feature Refinement (AFR) обрабатывает эти выбранные сигналы и перенастраивает их веса, усиливая последовательные и значимые паттерны и дополнительно подавляя шум. Наконец, сверточная нейронная сеть (CNN), обычно применяемая в анализе изображений, исследует локальные взаимодействия уточнённых признаков, улавливая тонкие взаимосвязи между группами генов, которые могут указывать на конкретный подтип заболевания или биологическое состояние.
Испытание модели
Фреймворк был протестирован на трёх крупных открытых ресурсах: наборе данных по раку молочной железы из The Cancer Genome Atlas, одно-клеточном наборе по меланоме из Gene Expression Omnibus и эпигеномном наборе из проекта ENCODE. В сумме эти коллекции включают тысячи образцов и десятки тысяч признаков на образец, охватывая активность генов и химические метки на ДНК. По всем наборам данных гибридная модель превзошла несколько современных подходов, улучшив точность и ключевые метрики качества классификации, такие как площадь под кривой ROC (AUC) и F1-метрика примерно на 5–8 процентных пунктов. Важно, что эти улучшения не шли в ущерб прозрачности: модель генерирует карты внимания от TabNet и карты активаций от CNN, которые показывают, какие гены и регионы были наиболее влиятельны в каждом прогнозе.
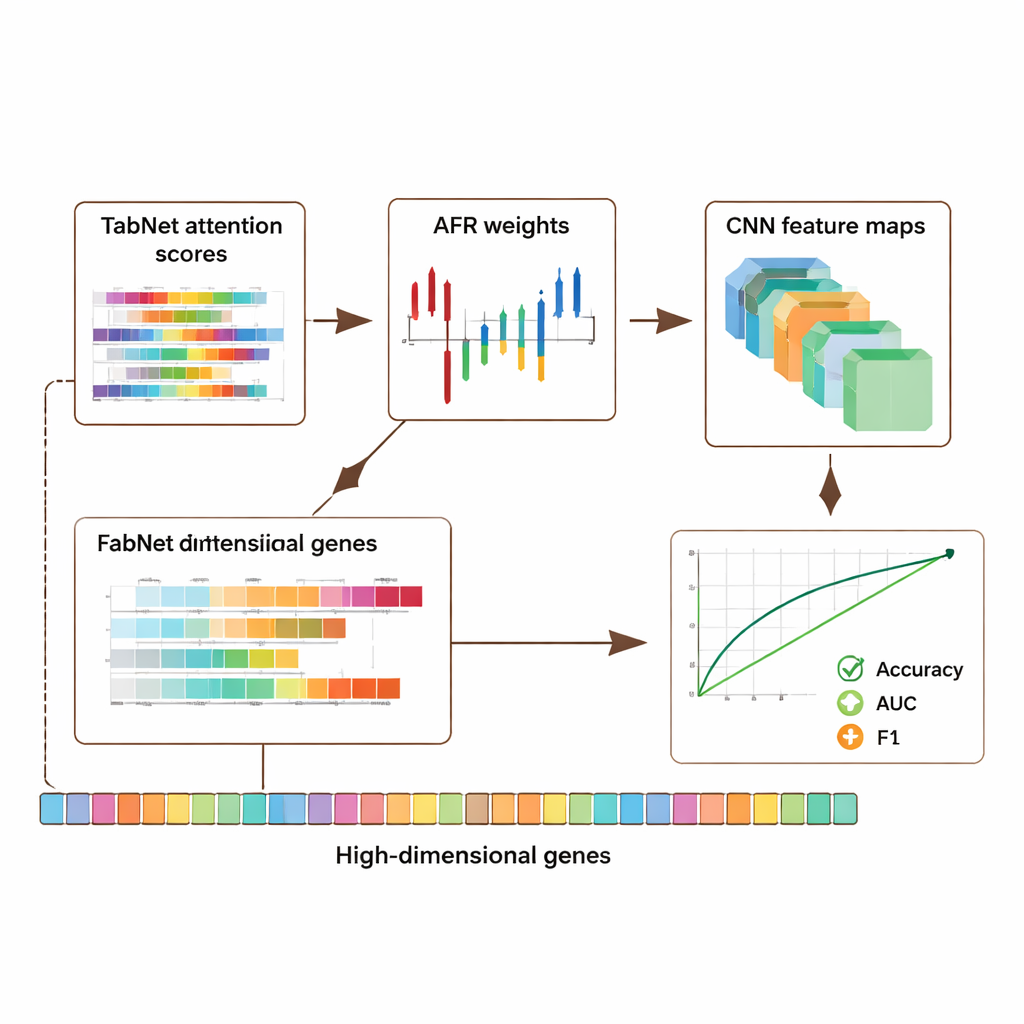
Баланс между точностью, приватностью и доверием
Поскольку геномные данные глубоко личные, авторы также изучили способы защиты приватности при сохранении полезного сигнала. Они предложили адаптивный механизм приватности, который добавляет больше шума к высокочувствительным признакам и меньше — к остальным, в сочетании с маскированием выбранных входов. Тесты показали, что даже при введении умеренного шума модель сохраняет высокую точность и способность различения, а производительность плавно снижается по мере ужесточения защиты. При этом интерпретируемые карты внимания и активаций часто указывали на гены, уже известные своей ролью в раке и регуляции иммунитета, что свидетельствует о том, что система не просто запоминает данные, но захватывает биологически значимые сигналы. Абляционное исследование — систематическое удаление частей архитектуры — подтвердило, что каждый модуль, особенно слой AFR, вносит измеримый вклад в производительность.
Что это значит для медицины будущего
Проще говоря, эта работа предлагает более умный способ просеивать огромные геномные таблицы в поисках паттернов, связанных с болезнью, одновременно показывая, какие записи в таблице были наиболее важны. Комбинируя целенаправленный выбор признаков, тщательную их доработку и распознавание паттернов, гибридная модель повышает точность прогнозов, остаётся вычислительно управляемой и предоставляет визуальные подсказки, которые клиницисты и биологи могут интерпретировать. Хотя необходимы дополнительные испытания на более широких и разнообразных группах пациентов, такие подходы могут помочь выявлять новые биомаркеры, уточнять подтипы заболеваний и поддерживать клинические инструменты в прецизионной медицине — приближая анализ ДНК с помощью ИИ к реальному использованию.
Цитирование: Swain, M.K., Kamila, N.K., Jena, L. et al. Hybrid deep learning framework for accurate classification of high dimensional genomic data. Sci Rep 16, 5919 (2026). https://doi.org/10.1038/s41598-026-36128-7
Ключевые слова: геномное глубокое обучение, обнаружение биомаркеров рака, интерпретируемый ИИ, персонализированная медицина, защита приватности в геномике