Clear Sky Science · ru
Количественное исследование цитотоксических соединений с использованием графовых дескрипторов и машинного обучения
Почему это исследование важно для будущих противораковых препаратов
Препараты против рака, убивающие опухолевые клетки и известные как цитотоксические средства, часто балансируют между спасением жизней и возникновением серьёзных побочных эффектов. Для разработки более безопасных и эффективных лекарств учёным нужны быстрые и надёжные методы предсказания того, как эти вещества перемещаются по организму — насколько хорошо они всасываются, как легко проходят через клеточные мембраны и где в итоге накапливаются. В этой работе показано, как математические описания молекул в сочетании с современными алгоритмами машинного обучения могут точно оценивать ключевое свойство, которое определяет такое поведение, что потенциально ускоряет поиск лучших противораковых препаратов.
Ключевая поверхность, которая контролирует, куда могут попадать препараты
Одна из центральных идей статьи — топологическая полярная площадь поверхности, или Top_PSA. Проще говоря, это число отражает, какая доля поверхности молекулы приходится на «полярные» участки — части, любящие воду и способные образовывать водородные связи. Молекулы с очень большой полярной площадью поверхности часто с трудом пересекают липидные мембраны и могут плохо всасываться при приёме внутрь. Молекулы с очень низкой полярной площадью поверхности слишком легко проходят многие барьеры, что иногда вызывает нежелательные эффекты в чувствительных тканях, например в мозге. Top_PSA стала популярным быстрым показателем для оценки этих транспортных свойств, поскольку её можно вычислить по 2D‑рисунку молекулы, не прибегая к медленным 3D‑симуляциям.
Преобразование молекулярных рисунков в числа
Исследователи собрали курируемый набор из 156 различных цитотоксических соединений, взятых из реальных противораковых препаратов и экспериментальных агентов. Затем каждую молекулу они преобразовали в 58 так называемых дескрипторов — чисел, которые отражают такие характеристики, как число атомов, количество колец, гибкость связей, число атомов, способных образовывать водородные связи, и степень полярности или электроотрицательности различных участков. Многие из этих дескрипторов основаны на теории графов, где молекула рассматривается как сеть узлов и связей. Этот насыщенный числовой портрет каждой молекулы послужил входными данными для компьютерных моделей, которые пытались предсказать значения Top_PSA, вычисленные с помощью широко используемых химических инструментов.
Испытание нескольких подходов к точному предсказанию
Чтобы найти наилучший способ связать дескрипторы с Top_PSA, команда сравнила несколько стратегий моделирования. Они пробовали стандартную линейную регрессию, а также две «регуляризованные» версии — гребневую (ridge) и LASSO-регрессию, которые лучше справляются с шумной и пересекающейся информацией. Также были изучены разные схемы предобработки данных: обучение моделей непосредственно на исходных дескрипторах, сжатие с помощью метода главных компонент (PCA), масштабирование, уменьшающее влияние экстремальных значений (robust scaling), корректировка выбросов и отбор с удалением сильнокоррелированных признаков с помощью показателя дисперсионного надувания (variance inflation factor). Каждый подход оценивался внимательно с использованием k‑кратной кросс‑валидации — метода, который многократно делит данные на обучающие и тестовые подмножества, чтобы защититься от переобучения.
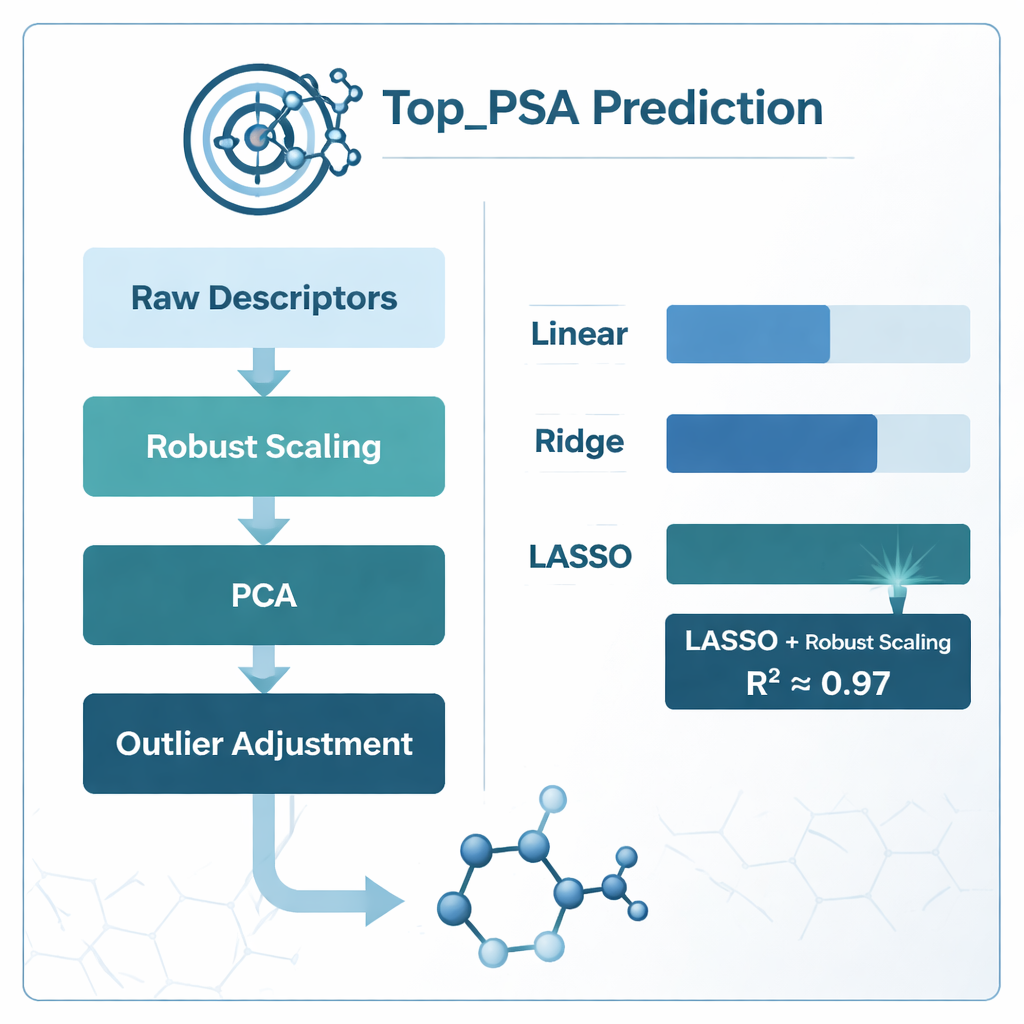
Что сработало лучше и чему научились модели
Ясным победителем стала комбинация robust scaling и LASSO-регрессии, которая показала коэффициент детерминации (R²) примерно 0,97 — то есть модель смогла объяснить примерно 97% вариации Top_PSA в наборе из 156 препаратов. Модели на основе PCA почти догнали по точности, но их труднее было интерпретировать химически, поскольку исходные дескрипторы смешивались в абстрактные компоненты. Простое удаление коррелированных дескрипторов с помощью variance inflation factor на самом деле ухудшало результаты, что говорит о том, что некоторые перекрывающиеся меры всё ещё содержат полезную химическую информацию. Анализ весов дескрипторов, сохранённых LASSO ненулевыми, показал, что самыми важными факторами являются наличие гетероатомов, таких как азот и кислород, способность отдавать или принимать водородные связи и индексы, отражающие распределение электроотрицательных атомов по молекулярному графу — все признаки, согласующиеся с интуитивным химическим пониманием полярной площади поверхности.
Как это может помочь в создании лучших лекарств
Для широкой аудитории ключевой вывод таков: аккуратно подготовленные математические отпечатки молекул в сочетании с правильно подобранными методами машинного обучения могут давать быстрые и надёжные оценки того, насколько «липкими» или «скользкими» будут противораковые препараты при движении по организму. Исследование даёт практические рекомендации другим учёным по предобработке дескрипторных данных, по выбору подходов к моделированию и по тому, каких упрощений следует избегать. В долгосрочной перспективе такие надёжные и интерпретируемые модели Top_PSA могут помочь химикам отфильтровывать огромные виртуальные библиотеки потенциальных препаратов, сосредотачивая усилия на соединениях с подходящим балансом прохождения через мембраны и безопасности — важный шаг к более эффективным и менее токсичным противораковым лечением.
Цитирование: Ahmad, S., Javed, S., Khalid, S. et al. A quantitative study of cytotoxic compounds using graph based descriptors and machine learning. Sci Rep 16, 5076 (2026). https://doi.org/10.1038/s41598-026-35728-7
Ключевые слова: цитотоксические препараты, полярная площадь поверхности, молекулярные дескрипторы, машинное обучение, проницаемость препаратов