Clear Sky Science · ru
Дисфункция митохондрий и нарушение регуляции Ca2+ в нейронах человека, полученных из iPSC, несущих мутацию пресенилина‑1, проявляются при стрессе через механизм, независимый от MCU‑1
Почему это важно для болезни Альцгеймера
Болезнь Альцгеймера часто описывают через липкие амилоидные бляшки в мозге, но задолго до провалов памяти «энергетические станции» внутри нейронов — митохондрии — и управление ионами кальция могут уже начать давать сбои. В этом исследовании использованы человеческие нейроны, выращенные из клеток кожи человека, несущего известную семейную мутацию Альцгеймера, чтобы задать простой, но ключевой вопрос: насколько рано и каким образом начинает нарушаться выработка энергии и кальциевый баланс?
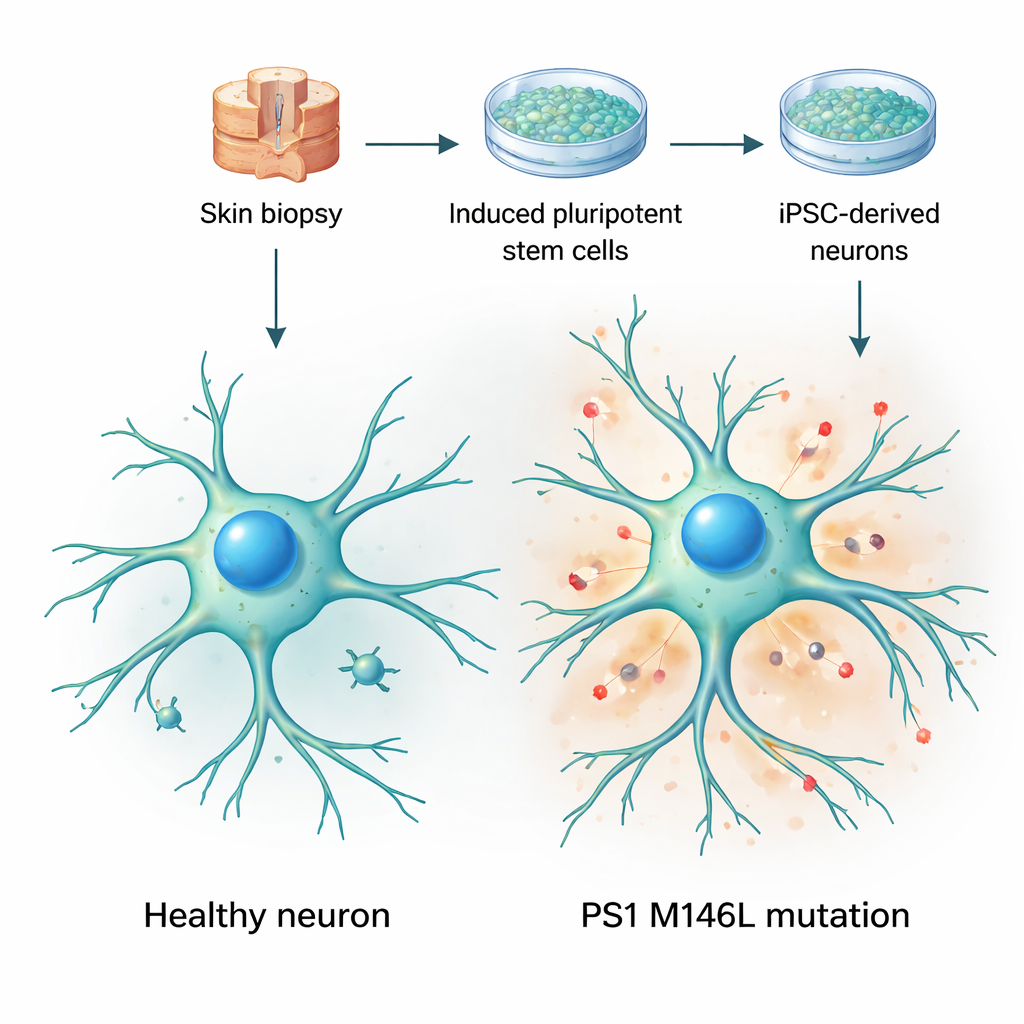
Превращение клеток кожи в живые модели мозга
Исследователи начали с биопсий кожи у двух женщин: одной здоровой доброволки и одной бессимптомной носительницы мутации пресенилина‑1 M146L, выявленной в аргентинской семье с ранним началом болезни Альцгеймера. Они перепрограммировали клетки кожи в индуцированные плюрипотентные стволовые клетки — клетки, способные превратиться практически в любую ткань — а затем направили их дифференцировку в нейроны. В течение нескольких недель в культуре эти клетки приобрели типичную нейронную морфологию, отросли длинные ветвящиеся процессы и стали экспрессировать стандартные нейронные маркеры. Важно, что и контрольные, и мутантные клетки созревали с похожей скоростью и выглядели в целом здоровыми, что позволило команде сосредоточиться на тонких функциональных изменениях, а не на явной гибели или повреждении клеток.
Электрические сигналы и кальций под нагрузкой
Нейроны зависят от строгого контроля кальция — заряженного атома, который действует как быстрый выключатель для многих клеточных процессов. С помощью флуоресцентных красителей команда отслеживала, как меняется уровень кальция в клетках при их электрической стимуляции калием или активации сигнальными молекулами. При простой деполяризующей стимуляции нейроны с мутацией M146L демонстрировали менее выраженные повышения кальция по сравнению с контролем, что указывает на проблемы с поддержанием электрических и ионных градиентов, которые обычно обеспечивают вход кальция. Однако когда экспериментаторы вызвали более стрессовую ситуацию — принудительный выход кальция из внутренних депо в эндоплазматическом ретикулуме — различия стали очевиднее. В ответ на этот стресс митохондрии мутантных нейронов захватывали заметно меньше кальция, чем митохондрии контрольных клеток, что указывает на сниженную способность буферизовать опасные всплески кальция.
Разъединение использования энергии и кальциевого баланса
Чтобы понять, как изменённый контроль кальция отражается на метаболизме клеток, исследователи измеряли потребление кислорода нейронами — прямой показатель митохондриальной активности. Удивительно, но нейроны с мутацией M146L «потребляли» больше: их базовый и максимальный уровни потребления кислорода, а также доля кислорода, связанная с синтезом АТФ, были выше, чем в контрольных клетках. При этом эффективность связывания потребления кислорода с производством АТФ выглядела схожей, и не было увеличения числа митохондрий или ключевых ферментов, отвечающих за синтез АТФ. Вместо этого митохондрии в мутантных нейронах были длиннее и более трубчатые, с повышенным уровнем белка слияния митофузина‑1 — паттерн, часто наблюдаемый при хроническом, низкоуровневом стрессе. Эти гиперактивные, удлинённые митохондрии также генерировали больше реактивных форм кислорода — нестабильных молекул, которые при недостаточном контроле могут повреждать белки и ДНК.
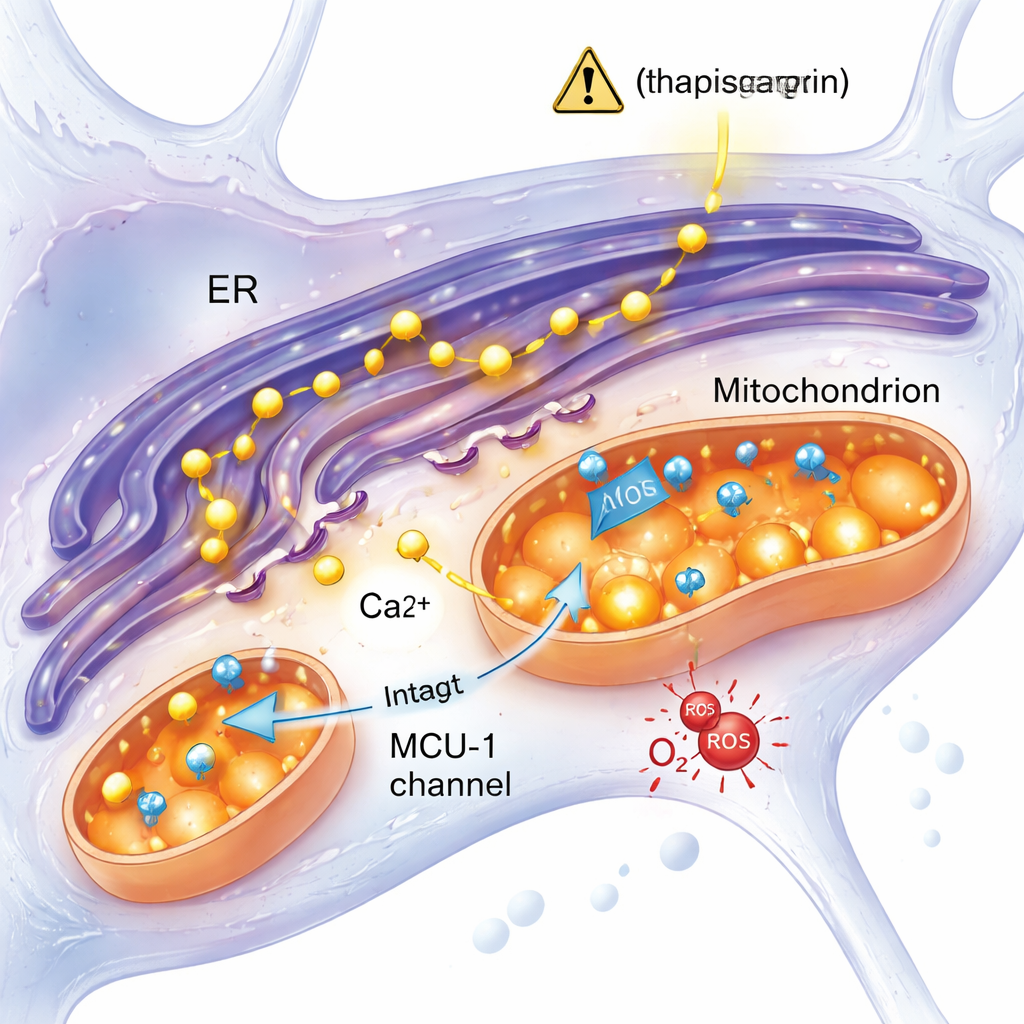
Стресс‑ответ, независимый от ключевого кальциевого канала
Одна из ведущих гипотез в исследованиях болезни Альцгеймера состоит в том, что избыток кальция из эндоплазматического ретикулума устремляется в митохондрии через канал, называемый митохондриальным uniporter’ом кальция (MCU‑1), перегружая их и вызывая дисфункцию. В этом исследовании эту идею проверяли напрямую. Когда команда блокировала MCU‑1 специфическим ингибитором, и контрольные, и мутантные нейроны демонстрировали резкое снижение захвата кальция митохондриями, что подтверждает работоспособность самого канала в обеих группах. Более того, когда выход кальция вызывался через более физиологический путь с участием IP3‑рецептора — ещё одного ключевого кальциевого клапана — мутантные и контрольные клетки отвечали схожим образом. Эти результаты отводят внимание от неисправного канала MCU‑1 и скорее указывают на то, что нарушены физические и функциональные контакты между эндоплазматическим ретикулумом и митохондриями или иные аспекты их взаимодействия в мутантных нейронах.
Что это значит для понимания и лечения болезни
В сумме полученные данные рисуют образ человеческих нейронов с мутацией PS1 M146L как клеток, которые в покое выглядят нормально, но реагируют ненормально под стрессом. Их митохондрии плохо захватывают кальций при внезапном высвобождении из внутренних депо, и в то же время работают активнее — потребляя больше кислорода и генерируя больше реактивных форм кислорода, будто застряв в энергозатратном компенсаторном режиме. Поскольку это наблюдается в живых нейронах человека до появления клинических симптомов, работа поддерживает идею, что нарушение кальциевой сигнализации и ранняя «перегрузка» митохондрий являются первоначальными событиями при Альцгеймере, а не только поздними следствиями. Для неспециалистов ключевой вывод таков: поддержание баланса между кальциевыми сигналами и митохондриальным энергопроизводством может быть столь же важно для профилактики болезни, как и воздействие на более известные амилоидные бляшки.
Цитирование: Wilson, C., Galeano, P., Remedi, M.M. et al. Mitochondrial dysfunction and Ca2+ dysregulation in human iPSC-derived neurons carrying presenilin-1 mutation arise under stress via an MCU-1-independent mechanism. Sci Rep 16, 6002 (2026). https://doi.org/10.1038/s41598-026-35597-0
Ключевые слова: болезнь Альцгеймера, митохондрии, кальциевый сигнал, мутация пресенилина‑1, нейроны, полученные из iPSC