Clear Sky Science · ru
Нарушение внутриклеточного гомеостаза железа через дисфункцию митохондрий, связанное с подавлением экспрессии ATP13A2
Почему важно железо внутри клеток мозга
Болезнь Паркинсона наиболее известна тремором и скованностью движений, но в глубине поражённых нейронов разворачивается другая история: железо, необходимый металл, начинает накапливаться там, где ему не место. В этом исследовании задаётся простая, но важная проблема: как именно происходит это накопление железа и каким образом оно вредит крошечным энергетическим станциям и перерабатывающим центрам в нервных клетках? Давая ответ, работа проливает свет на причины дегенерации определённых областей мозга при болезни Паркинсона и родственных расстройствах и указывает на новые подходы к лечению, выходящие за рамки простой замены дофамина.
Внимательнее к редкой генетической подсказке
Исследователи сосредоточились на редкой наследственной форме болезни Паркинсона, называемой PARK9, вызванной дефектами в гене ATP13A2. Этот ген кодирует белок, локализующийся в лизосомах — отделениях клетки, отвечающих за утилизацию и переработку. У людей с мутациями ATP13A2 также может развиваться состояние с отложениями железа в мозге. Эта связь сделала ATP13A2 удобной точкой входа для изучения нарушения баланса железа. Используя клеточную линию, имитирующую человеческие нейроны и сверхэкспрессирующую парковые белки альфа‑синуклеин, команда применяла короткие РНК для понижения уровня ATP13A2 и отслеживала изменения в распределении железа, энергетическом статусе и состоянии клеток.
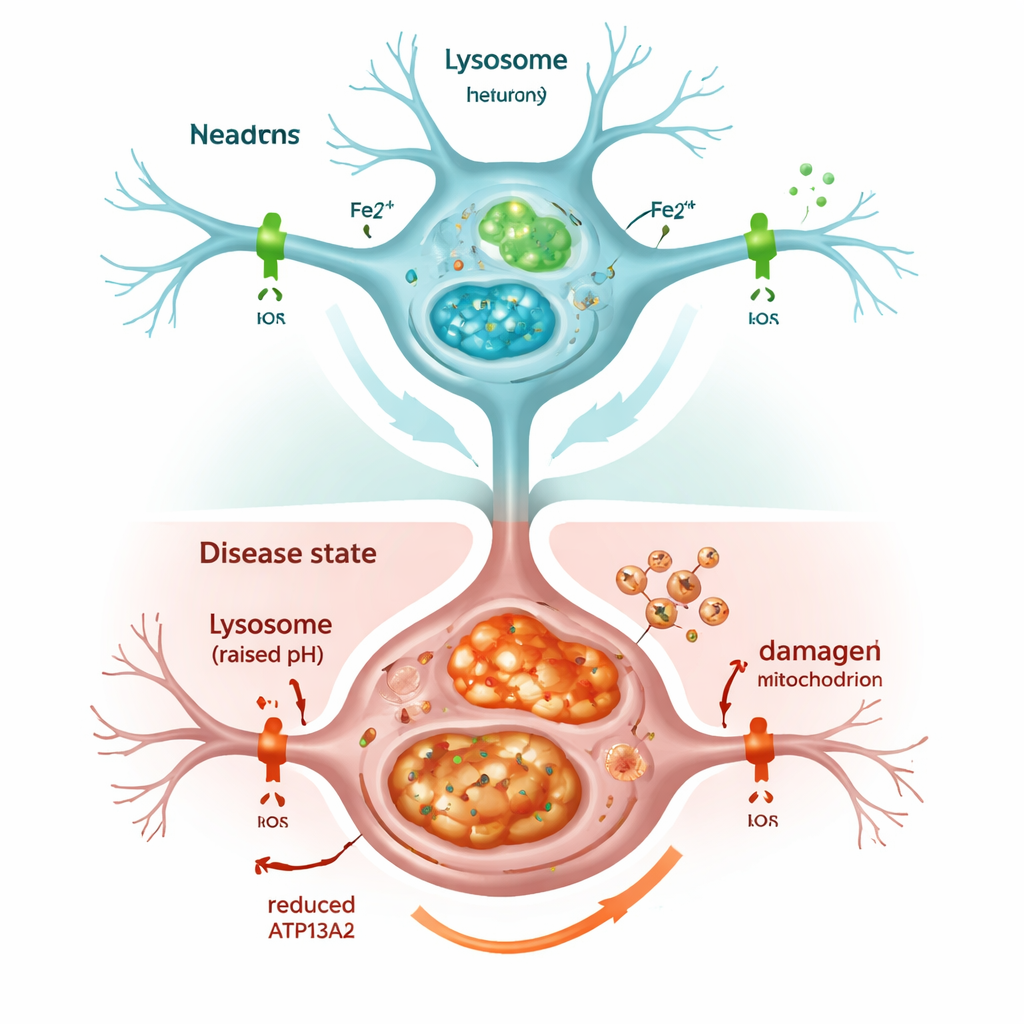
Когда система переработки клетки даёт сбой
Подавление ATP13A2 быстро ослабляло функцию лизосом. Их внутренняя кислотность, критичная для расщепления ненужного материала, падала, а маркеры аутофагии — процесса очистки клетки — накапливались вместо того, чтобы утилизироваться. В результате накапливался альфа‑синуклеин, что повторяет наблюдаемое при болезни Паркинсона. Клетки также демонстрировали общее повышение содержания железа, особенно химически активной формы Fe2+, внутри как лизосом, так и митохондрий. Клетка отвечала усилением производства ферритина, белка, запасающего железо, но этого оказалось недостаточно: перегруженные митохондрии вырабатывали избыток реактивных кислородных молекул, и выживаемость клеток снижалась. Лечение клеток железосвязывающим препаратом, похожим на клинически используемые, снизило этот окислительный стресс и частично восстановило жизнеспособность, подчёркивая, что избыток железа сам по себе был ключевым фактором повреждения.
Датчики железа перестают «слушать» металл
Обычно в клетке действует система обратной связи, замечающая повышение уровня железа и снижающая его поглощение. Белок IRP2 чувствует железо отчасти через сигнал, зависящий от гема, поступающий из митохондрий, и затем регулирует синтез белков‑транспортеров железа на поверхности клетки. В клетках с дефицитом ATP13A2 эта защита дала сбой. Транспортеры, ввозящие железо в клетку, оставались высокоэкспрессированными, хотя железо и так было повышено. Уровни белка IRP2 почти не менялись, и добавление внешнего железа не вызывало его привычного расщепления. Команда проследила причину до митохондрий: повреждённые митохондрии дышали менее эффективно, демонстрировали признаки нарушённого контроля качества (митофагии) и, что важно, утратили способность синтезировать гем — молекулу, содержащую железо, помогающую IRP2 фиксировать избыток железа. При недостатке гема IRP2 не получал «сигнал о переизбытке» и позволял дальнейший приток железа.
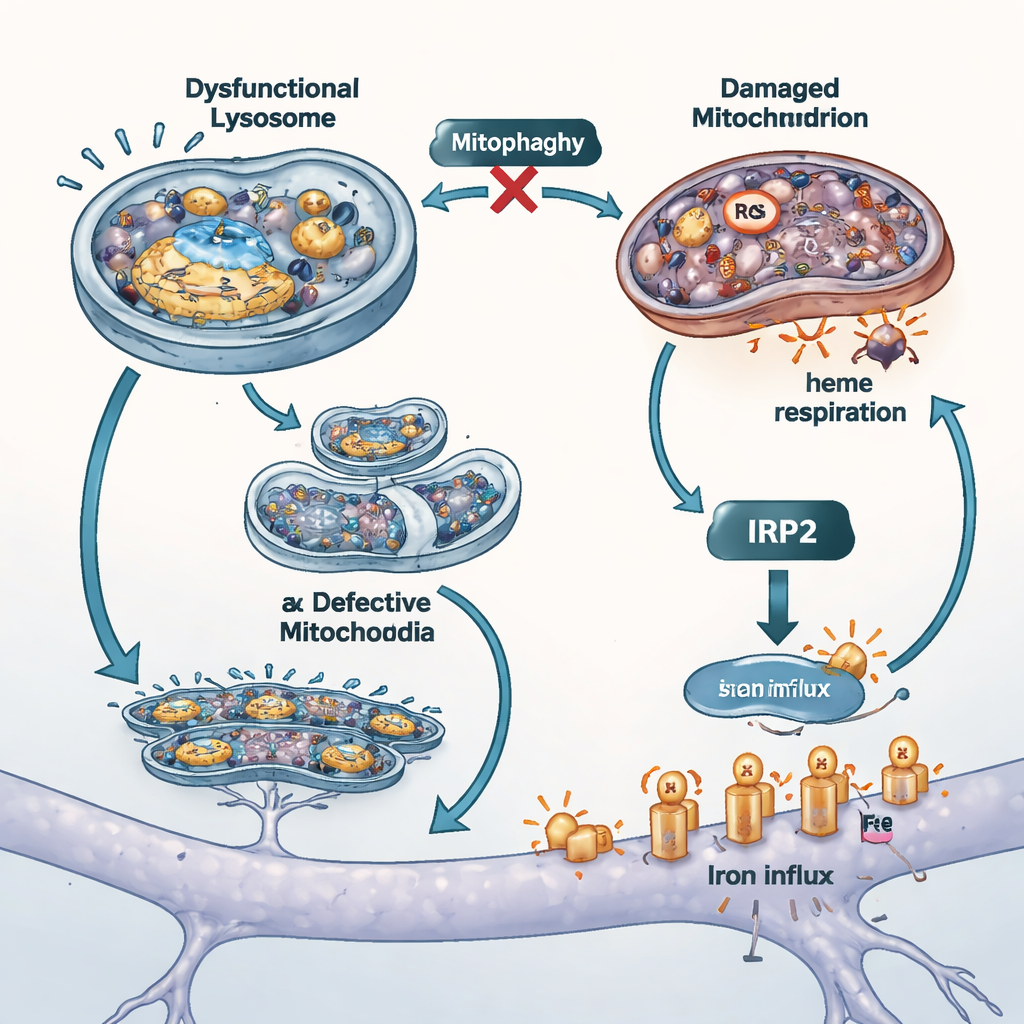
Перекрытие поступления железа и испытания в других моделях
Чтобы оценить вклад неконтролируемого притока железа в повреждение клеток, учёные заблокировали два основных пути его поступления. Они использовали апотрансферрин (трансферрин без железа) для конкуренции за один из переносчиков и малое вещество, ослабляющее активность другого переносчика — DMT1. Оба подхода снизили общее и свободное железо в клетках, уменьшили митохондриальный оксидативный стресс и улучшили выживаемость, что говорит о том, что поверхностные каналы для железа являются важными усилителями повреждения при потере ATP13A2. Исследователи также повторили ключевые эксперименты в клетках, лишённых другого связанного с болезнью Паркинсона гена PINK1, известного нарушениями митофагии. Эти клетки показали ту же комбинацию накопления железа и ослабленного синтеза гема, подтверждая идею о тесной взаимосвязи контроля качества митохондрий и баланса железа при разных формах болезни.
Что это означает для болезни Паркинсона и будущих подходов к лечению
Проще говоря, исследование описывает порочный круг. При подавлении ATP13A2 лизосомы перестают эффективно убирать повреждённые компоненты, включая дефектные митохондрии. Ослабленные митохондрии начинают вырабатывать меньше энергии и меньше гема, что притупляет систему клеточного учёта железа. Железо продолжает поступать через поверхностные транспортеры, накапливается в уязвимых компартментах и запускает токсичные реакции, которые ещё сильнее повреждают митохондрии. Со временем этот цикл может объяснять, почему определённые нейроны гибнут при болезни Паркинсона и родственных нарушениях с накоплением железа. Результаты указывают, что будущие терапии могли бы не только удалять избыток железа, но и восстанавливать функцию лизосом, контроль качества митохондрий и синтез гема — воздействуя на причину проблемы, а не только устраняя металл постфактум.
Цитирование: Murakami, T., Ohuchi, K., Kiuchi, M. et al. Disruption of intracellular iron homeostasis through mitochondrial dysfunction associated with suppression of ATP 13A2 expression. Sci Rep 16, 5007 (2026). https://doi.org/10.1038/s41598-026-35368-x
Ключевые слова: Болезнь Паркинсона, железо в мозге, митохондрии, лизосомы, синтез гемa