Clear Sky Science · ru
AF2BIND: предсказание участков связывания малых молекул с использованием парного представления AlphaFold2
Поиск лекарственных целей в океане белков
Современные лекарства часто действуют, прикрепляясь к крошечным углублениям и щелям на поверхности белков внутри наших клеток. И даже при наличии огромных современных каталогов структур белков по-прежнему удивительно трудно заранее определить, куда именно может присоединиться малые молекула — потенциальный препарат. В этом исследовании представлен AF2BIND, простой, но мощный вычислительный инструмент, который извлекает информацию из внутренних слоёв AlphaFold2, прорывного предиктора структуры белков, чтобы выделять вероятные участки связывания лекарств в тысячах белков человека. Его цель — сузить поиск новых лекарств и выявить скрытые функциональные «горячие точки», которые упускают традиционные методы.
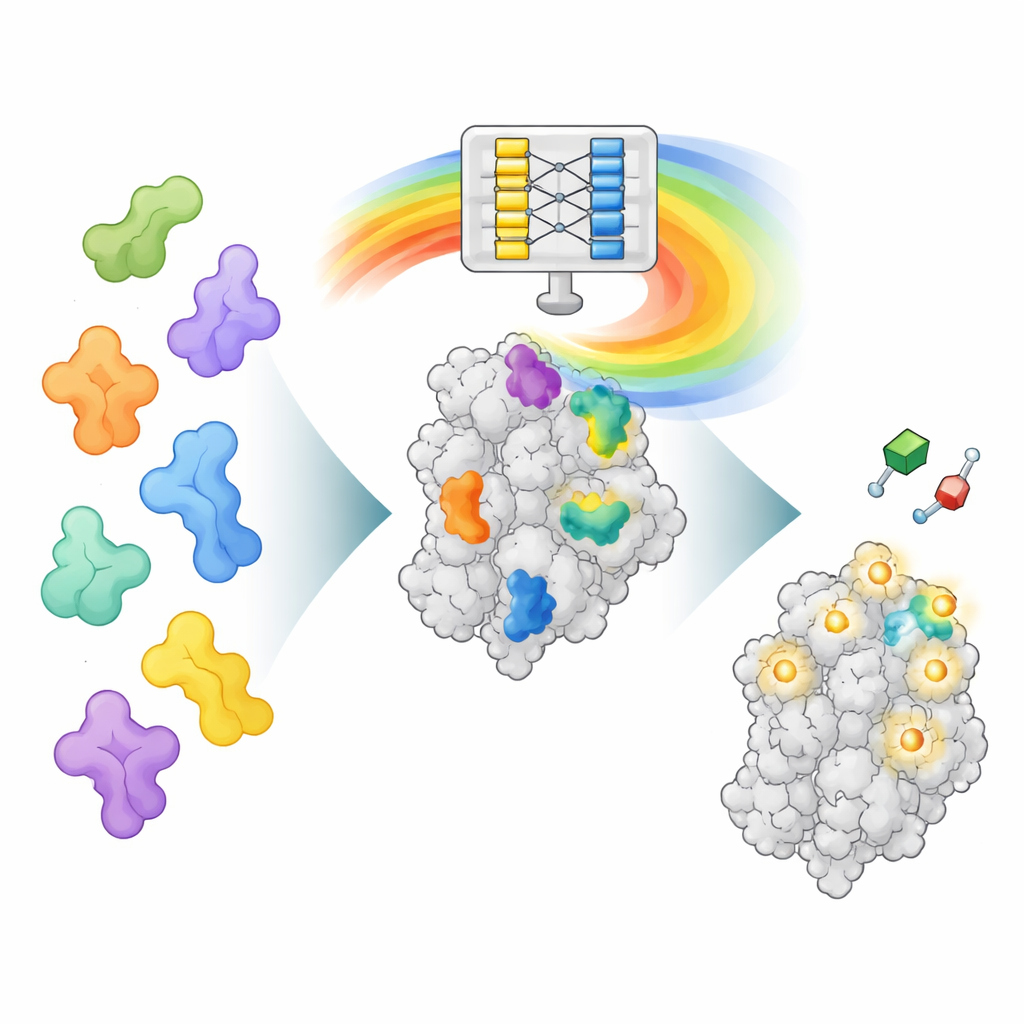
Новый способ читать «ум» AlphaFold
AlphaFold2 обучался предсказывать, как цепочка аминокислот складывается в трёхмерную структуру белка, а не искать места связывания лекарств. Однако, в процессе обучения сворачивания белков он также освоил богатые закономерности о том, как разные части белков взаимодействуют. AF2BIND обращается к одному из этих внутренних слоёв данных, называемому парным представлением, которое кодирует взаимосвязь каждой пары аминокислотных позиций в пространстве. Авторы подают в AlphaFold2 последовательность белка вместе с его каркасной структурой и добавляют 20 дополнительных аминокислот, по одной каждого типа, в виде отдельных «приманочных» цепочек. AlphaFold2 затем вычисляет, как белок взаимодействует с каждой приманочной остатком. Эти шаблоны взаимодействий становятся входными данными для очень простого логистического регрессионного моделирования, которое оценивает для каждой позиции белка вероятность того, что она принадлежит участку связывания малой молекулы.
Преобразование скрытых сигналов в практические предсказания
Обучение AF2BIND потребовало тщательно подобранного набора примерно из 1900 структур белок–лигандов, где малые молекулы связывались с высококачественными экспериментальными доказательствами. Исследователи приложили значительные усилия, чтобы избежать «жульничества» через сходство: они разделили данные так, чтобы тестовые белки не имели общей складки, последовательности или даже формы кармана связывания с теми, что использовались при обучении. На этом строгом бенчмарке парное представление AF2 превзошло несколько альтернативных встраиваний нейронных сетей, включая те, что основывались только на последовательности или на дизайне последовательности, обусловленном структурой. Используя только парные признаки, AF2BIND восстановил примерно две трети известных связывающих остатков среди верхнеранжированных предсказаний и показал высокую эффективность по стандартным метрикам классификации, оставаясь при этом робастным к умеренным изменениям формы белка и ориентации боковых цепей.
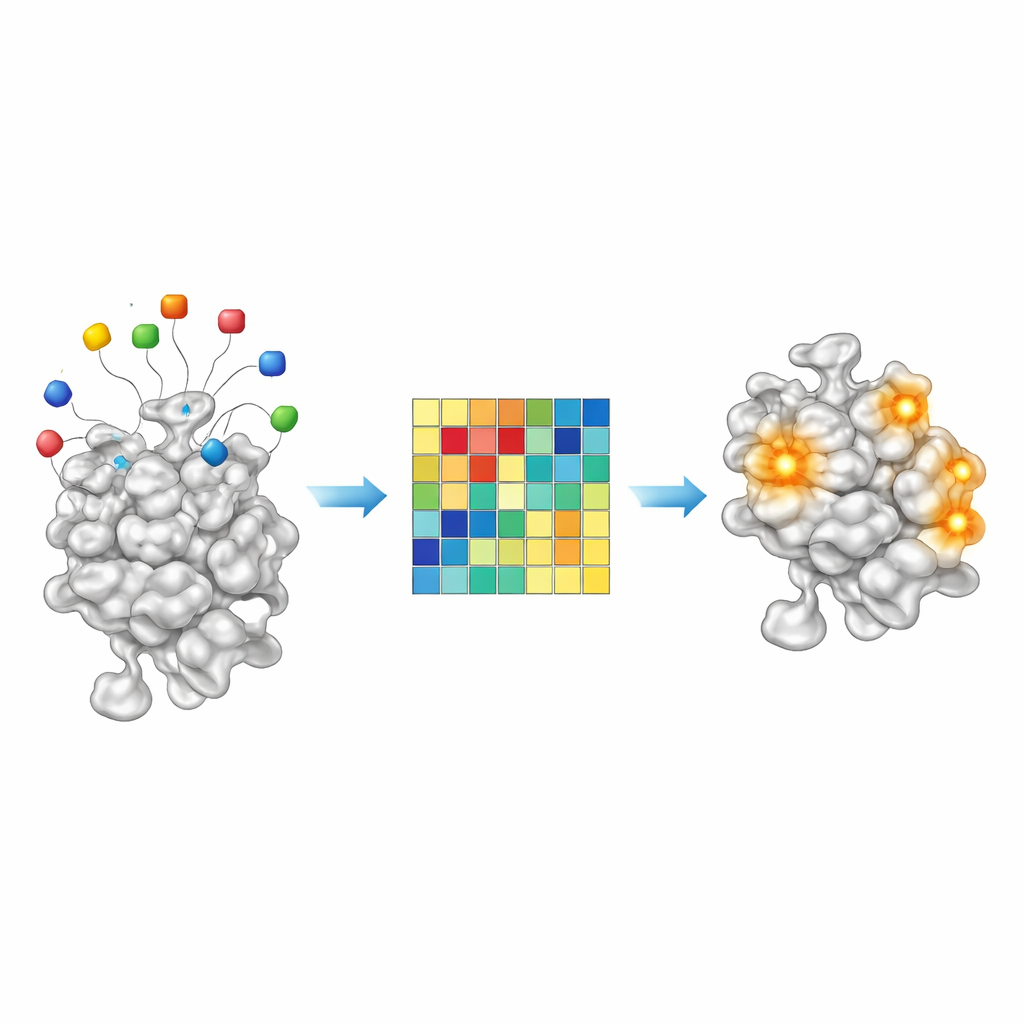
Чтение химических подсказок от приманочных остатков
Поскольку AF2BIND — это простой линейный модель, её решения необычно прозрачны для современной системы ИИ. Каждый из 20 приманочных аминокислот вносит измеримый вклад в итоговую оценку связывания для данной позиции белка. Изучая эти вклады в примерно 2000 комплексов белок–лиганд, авторы обнаружили, что определённые комбинации приманок сильнее активируются для жирных, углеродсодержащих лигандов, тогда как другие реагируют на более полярные, гидрофильные молекулы. Иными словами, паттерн активации приманок действует как грубая химическая «отпечаток пальца», указывающий, какие типы малых молекул предпочитает данный карман. Это наводит на мысль, что в будущем подходы типа AF2BIND могли бы не только отмечать, где может связываться препарат, но и намекать на ту химию, которая лучше всего подойдёт.
Сканирование человеческого протеома в поисках новых карманов
Вооружившись обученной моделью, команда затем запустила AF2BIND на предсказанных AlphaFold структурах всего человеческого протеома. Отсечив регионы с низкой уверенностью и разделив очень большие белки на управляемые структурные фрагменты, они сгруппировали близко расположенные высокооцененные остатки в кандидатные участки связывания. AF2BIND предсказал более 20 000 таких участков в более чем 13 000 белков. Поразительно, что большинство из них не перекрывалось с карманами, определёнными методами на основе гомологии, такими как AlphaFill, которые копируют лиганды из родственных кристаллических структур, ни с широко используемым детектором карманов P2Rank. Многие участки, найденные только AF2BIND, более мелкие или более размытые, чем классические скрытые карманы, и часто совпадают с областями, связывающими пептиды, РНК, ДНК или другие белки — интерфейсами, которые тем не менее могут быть нацелены малыми молекулами.
Последствия для поиска лекарств и понимания болезней
Чтобы оценить, насколько перспективны эти недавно предложенные участки для дизайна лекарств, авторы использовали независимый инструмент, который оценивает «лекарственность» на основе размера кармана, степени его замкнутости и химического окружения. В среднем участки AF2BIND получили оценки выше обычного порога привлекательности для лекарственных целей, включая те, что найдены в белках, связанных с наследственными заболеваниями. При сопоставлении с химопротеомными экспериментами, маркирующими реактивные цистеины в клетках, AF2BIND и P2Rank вместе объясняли почти половину наблюдаемых лигандообязуемых регионов, причём каждый метод выявлял случаи, которые другой пропускал. Работа демонстрирует, что внутренние представления, изученные сетями для предсказания структуры, можно перепрофилировать для массового картирования вероятных участков связывания лекарств, без предварительных знаний о каком-либо конкретном лигандe. Для неспециалистов ключевое сообщение в том, что те же прорывы в ИИ, которые предсказывают формы белков, начинают показывать, где и как лекарства могут лучше всего захватывать эти формы — что потенциально ускоряет поиск новых терапий и освещает ранее скрытые точки контроля в наших белках.
Цитирование: Gazizov, A., Lian, A., Goverde, C. et al. AF2BIND: predicting small-molecule binding sites using the pair representation of AlphaFold2. Nat Methods 23, 626–635 (2026). https://doi.org/10.1038/s41592-026-03011-2
Ключевые слова: участки связывания белков, поиск лекарств, AlphaFold2, вычислительная биология, структурная биоинформатика