Clear Sky Science · ru
Вариабельность скоростей и повторяющиеся ошибки последовательностей в филогенетике масштаба пандемии
Почему это важно для будущих вспышек
Когда новый вирус распространяется по всему миру, учёные спешат расшифровать его генетический код и восстановить его «родословное дерево». Такие деревья помогают отслеживать, как появляются варианты, как быстро они распространяются и работают ли меры контроля. Но во время COVID‑19 лаборатории так быстро секвенировали миллионы геномов SARS‑CoV‑2, что скрытые ошибки и особенности данных начали искажать картину. В этой работе представлены новые методы очистки и интерпретации таких огромных генетических наборов данных, которые дают более ясное представление о том, как вирус пандемии действительно эволюционирует и распространяется в популяциях.
Сложность работы с миллионами геномов
Геномная эпидемиология превращает геномы вирусов в практическую информацию для решений в области общественного здоровья. Для SARS‑CoV‑2 в мировое сообщество было передано более 20 миллионов геномов. Традиционные эволюционные инструменты создавались для более скромных задач, например для сравнения генов между видами, а не для обработки миллионов почти идентичных вирусных последовательностей, поступающих в реальном времени. На таком масштабе особенно проблемными становятся два фактора. Во‑первых, некоторые участки вирусного генома мутируют гораздо чаще, чем другие, что может сделать несвязанные вирусы внешне похожими. Во‑вторых, повторяющиеся технические ошибки в секвенировании и обработке данных могут имитировать настоящие мутации. Оба эффекта создают «ложные эхо» в дереве эволюции, порождая неясности в том, каким ветвям и группировкам можно доверять.
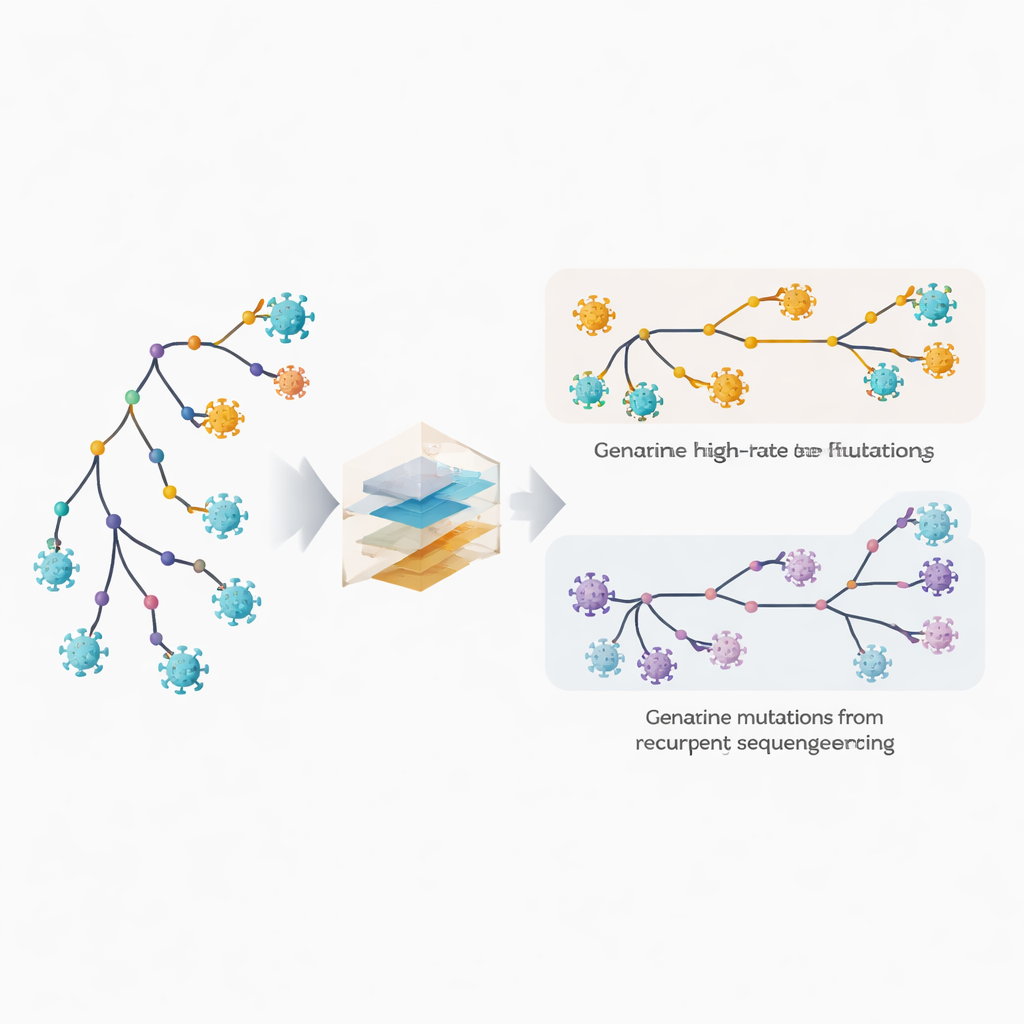
Выявление быстро меняющихся участков и скрытых ошибок
Авторы расширяют своё филогенетическое ПО MAPLE моделями, которые рассматривают каждую позицию в вирусном геноме как обладающую собственной динамикой. Вместо предположения о нескольких средних скоростях мутаций метод оценивает отдельную скорость для каждой позиции, используя огромное количество доступных геномов. Одновременно он допускает для каждой позиции собственную вероятность повторяющейся ошибки секвенирования или принятия консенсуса. Ключевой приём — сравнение частоты появления изменений на глубоких внутренних ветвях дерева, отражающих более старые, общие события, и на внешних кончиках, соответствующих отдельным геномам. Истинные биологические мутации распределены между внутренними и терминальными ветвями, тогда как технические ошибки проявляются преимущественно на кончиках. Используя эту закономерность, метод способен отделять подлинную быструю эволюцию от повторяющихся артефактов.
Более быстрые алгоритмы для густонаселённого дерева жизни
Обработка миллионов геномов обычно потребовала бы огромных вычислительных ресурсов. Чтобы сделать анализ практичным, команда переработала то, как MAPLE хранит и обновляет информацию о последовательностях на дереве. Вместо сравнения каждого генома с одним фиксированным референсом программное обеспечение выбирает «локальные референсы» внутри дерева и записывает близкие геномы как различия относительно этих опор. Такое компактное представление ускоряет сравнения между удалёнными частями дерева. Дополнительные улучшения касаются добавления новых образцов в уже существующее дерево, настройки длин ветвей и исследования вероятных альтернативных форм дерева, с опцией выполнения самых тяжёлых шагов параллельно на нескольких ядрах процессора.
Тестирование метода и очистка реальных данных
Чтобы проверить работу моделей, авторы сначала создали реалистичные симулированные наборы данных SARS‑CoV‑2 с известными паттернами мутаций и встроенными ошибками последовательностей. В этих тестах новый подход восстанавливал более правдоподобные эволюционные деревья и с высокой точностью обнаруживал индивидуальные ошибки, особенно при включении десятков тысяч геномов и более. Затем они обратились к реальным данным, проанализировав миллионы последовательностей SARS‑CoV‑2, для которых были доступны сырые риды. Сравнивая два разных конвейера построения консенсуса, они выявили конкретные позиции генома, неоднократно затрагиваемые артефактами, такими как проблемы связывания праймеров или смещение при вызове относительно референса. Подозрительные участки были замаскированы для дальнейшего анализа, а геномы с признаками контаминации или смешанной инфекции были отфильтрованы, что привело к курированному выравниванию более чем двух миллионов высококачественных последовательностей.
Более чёткая глобальная картина родословного дерева вируса
Используя очищенный набор данных, авторы реконструировали глобальное филогенетическое дерево SARS‑CoV‑2 и отобразили взаимосвязи основных вариантов. Их дерево иногда предлагает тонко отличающиеся отношения по сравнению с предыдущими публичными деревьями, часто такими, которые требуют меньше мутационных событий и лучше соответствуют статистической модели. Рамки метода также подчёркивают места, где метки линий могут быть несогласованы с основной генетической историей, указывая на возможные рекомбинанты или проблемные геномы для более тщательной проверки. Хотя некоторые сложности остаются — например, переобучение при скудных данных или влияние сильно контаминированных образцов — работа демонстрирует, что теперь возможно строить более надёжные эволюционные деревья в масштабе пандемии. Для неспециалиста итог прост: лучшее обращение с ошибками и «горячими точками» мутаций даёт более чёткое понимание того, как патогены распространяются и изменяются, помогая учёным и здравоохранительным службам реагировать быстрее и с большей уверенностью при будущих вспышках.
Цитирование: De Maio, N., Willemsen, M., Martin, S. et al. Rate variation and recurrent sequence errors in pandemic-scale phylogenetics. Nat Methods 23, 565–573 (2026). https://doi.org/10.1038/s41592-025-02932-8
Ключевые слова: геномика SARS‑CoV‑2, филогенетические методы, ошибки секвенирования, вариация скорости мутаций, геномная эпидемиология