Clear Sky Science · ru
Переносимые модели энантиоселективности из разреженных данных
Более умный способ найти подходящий катализатор
Химики часто ищут лучшие лекарства и материалы, пытаясь связывать атомы углерода в очень конкретные трехмерные перестановки. Добиться тонкой «правозеркальной» или «левозеркальной» конфигурации — известной как энантиоселективность — обычно означает перебор множества металлических катализаторов и условий реакции методом проб и ошибок. В этой работе предложен подход, который использует относительно небольшие объемы экспериментальных данных в сочетании с быстрыми компьютерными расчетами, чтобы предсказывать, какие никелевые катализаторы дадут желаемую хиральность в широком диапазоне реакций, что потенциально экономит химикам недели или месяцы работы в лаборатории.
Почему управлять хиральными молекулами так сложно
Многие лекарства и природные продукты существуют в виде зеркальных форм, которые в организме могут вести себя очень по-разному. Катализаторы, отдающие предпочтение одной зеркальной форме перед другой, поэтому невероятно ценны. Но проектировать такие катализаторы сложно. Традиционная квантовая химия теоретически способна вычислить, какой путь реакции предпочтительнее, однако даже крошечные погрешности в энергии приводят к большим ошибкам в прогнозах селективности, а сами расчеты медленны. Проще статистические модели, напротив, быстры, но часто игнорируют тонкое взаимодействие между металлическим катализатором и реагирующими молекулами, особенно когда механизм реакции может изменяться при смене реагентов.
Фиксация ключевых моментов в реакции
Авторы преодолевают этот разрыв, сосредоточившись на наиболее критических стадиях никелевого перекрестного присоединения: шагах, где образуются новые углерод–углеродные связи, и стадии релиза конечного продукта. Вместо дорогостоящих высокоточных симуляций они используют упрощенный квантовый метод для построения трехмерных структур ключевых переходных состояний и интермедиатов для множества возможных комбинаций катализаторов и субстратов. Из этих структур они извлекают сотни физически значимых дескрипторов, таких как степень загруженности области вокруг определенных атомов или легкость движения электронов. Эти числовые характеристики затем подаются в простые модели линейной регрессии, связывающие структурные признаки с измеренной селективностью.
Обучение на разреженных данных для направления новых экспериментов
Ключевое достижение работы — умение максимально эффективно использовать разреженные данные — ограниченное число комбинаций катализаторов и субстратов, как правило описываемых в статье. В одном из тематических исследований команда повторно рассматривает никелевую реакцию, соединяющую стироловые оксиды с арил-йодидами. Они показывают, что дескрипторы, полученные из наиболее релевантного переходного состояния, превосходят те, которые извлечены из упрощенных фрагментов катализатора, несмотря на меньшую стоимость вычислений. Имея такие модели, они виртуально тестируют гораздо больше лигандов на известных парах субстратов и выявляют новые варианты катализаторов, которые повышают энантомерный избыток в особенно упорных примерах, при этом избегая десятков ненужных экспериментов.
Перенос знаний между разными реакциями
Подход мощен тем, что его можно переносить на разные, но близкие по механизму никелевые реакции. В другом наборе исследований авторы объединяют данные нескольких типов никелевых реакций, которые все формируют связи между sp3-гибридизированными атомами углерода и партнерами вроде арильных или алкенильных групп, даже если точные условия или реагенты различаются. Строя модели на основе одних и тех же механистически значимых дескрипторов, они успешно предсказывают энантиоселективность для новых лигандов, новых комбинаций субстратов и даже для совершенно нового класса реакций образования углерод–углеродной связи, не входивших в обучающую выборку. Анализ наиболее значимых дескрипторов также указывает на то, на каком этапе каталитического цикла устанавливается хиральность для каждого семейства реакций.
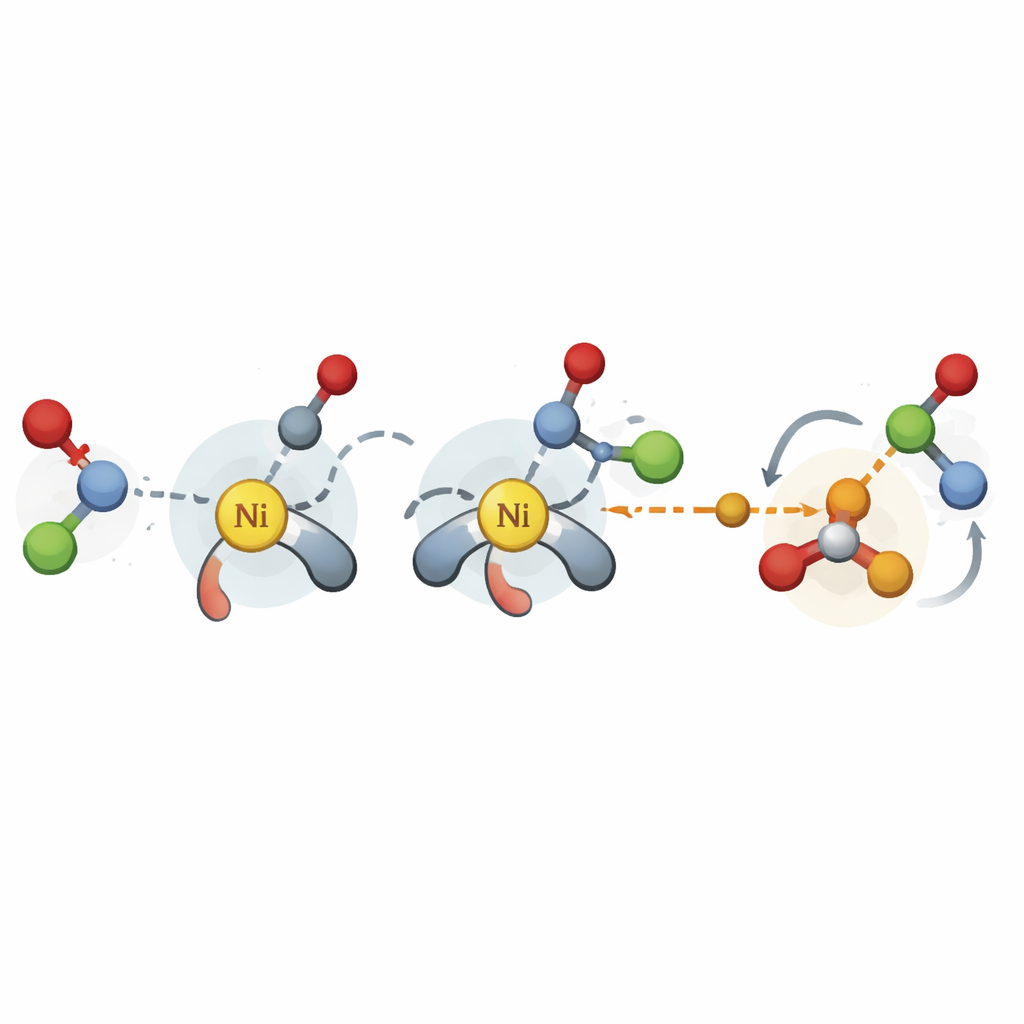
Помощь химикам в более быстром запуске новых реакций
В финальной демонстрации авторы используют свою схему дескрипторов вместе с платформой байесовской оптимизации для разработки никелевого соединения бензилизованных ацеталей и арил-йодидов, которое ранее не развивалось в асимметрическом направлении. Начиная с литературных данных по другим реакциям, модель рекомендует небольшие наборы перспективных лигандов для тестирования, быстро сужая поиск до наилучшего класса всего за несколько десятков экспериментов. Для химика это означает практический инструмент для «холодного старта» нового каталитического проекта: накопив несколько начальных результатов, модель может предложить, какие хиральные лиганды с наибольшей вероятностью обеспечат высокую энантиоселективность. В целом исследование показывает, что продуманно подобранные и недорогие вычислительные признаки способны превратить ограниченные прошлые данные в широко полезное руководство по созданию нового поколения селективных реакций.
Цитирование: Gallarati, S., Bucci, E.M., Doyle, A.G. et al. Transferable enantioselectivity models from sparse data. Nature 651, 637–646 (2026). https://doi.org/10.1038/s41586-026-10239-7
Ключевые слова: асимметричный катализ, никелевое перекрестное соединение, машинное обучение в химии, оптимизация реакций, прогнозирование энантиоселективности