Clear Sky Science · ru
Двусторонние CRISPR-скрининги расшифровывают зависимую от GLIS3 фибротическую клеточную сеть
Когда заживление превращается в вредный рубец
Наш кишечник предназначен для самовосстановления после каждогo повреждения и раздражения. Но при хронических заболеваниях, таких как болезнь Крона и язвенный колит, этот процесс заживления может пойти не так, в результате образуется плотная, жесткая рубцовая ткань, которая сужает просвет и может потребовать хирургического вмешательства. В этом исследовании раскрывается скрытый диалог между иммунными и структурными клетками кишечника, который приводит к такому рубцеванию, и выявляется ключевой переключатель — ген GLIS3, который может дать новую возможность разорвать этот порочный круг.
Скрытая сеть в воспалённом кишечнике
Чтобы понять, почему у некоторых пациентов развивается упорное воспаление и фиброз (рубцевание), исследователи создали клеточный «атлас» человеческого кишечника. Они объединили одноклеточный РНК-секвенсинг, считывающий активные гены в отдельных клетках, со пространственным профилированием, которое отображает, где эти клетки расположены в реальных тканевых срезах. Используя образцы от людей с болезнью Крона, язвенным колитом и контрольных доноров, они картировали более четырёх миллионов клеток по всей толщине кишечной стенки. Среди этого множества выделилась одна подгруппа фибробластов: ассоциированные с воспалением фибробласты (IAF). Эти клетки скапливались в областях активного и хронического колита и несли генетический отпечаток, связанный с устойчивостью к стандартной противо-TNF терапии, что указывает на их центральную роль в трудно поддающемся лечению заболевании. 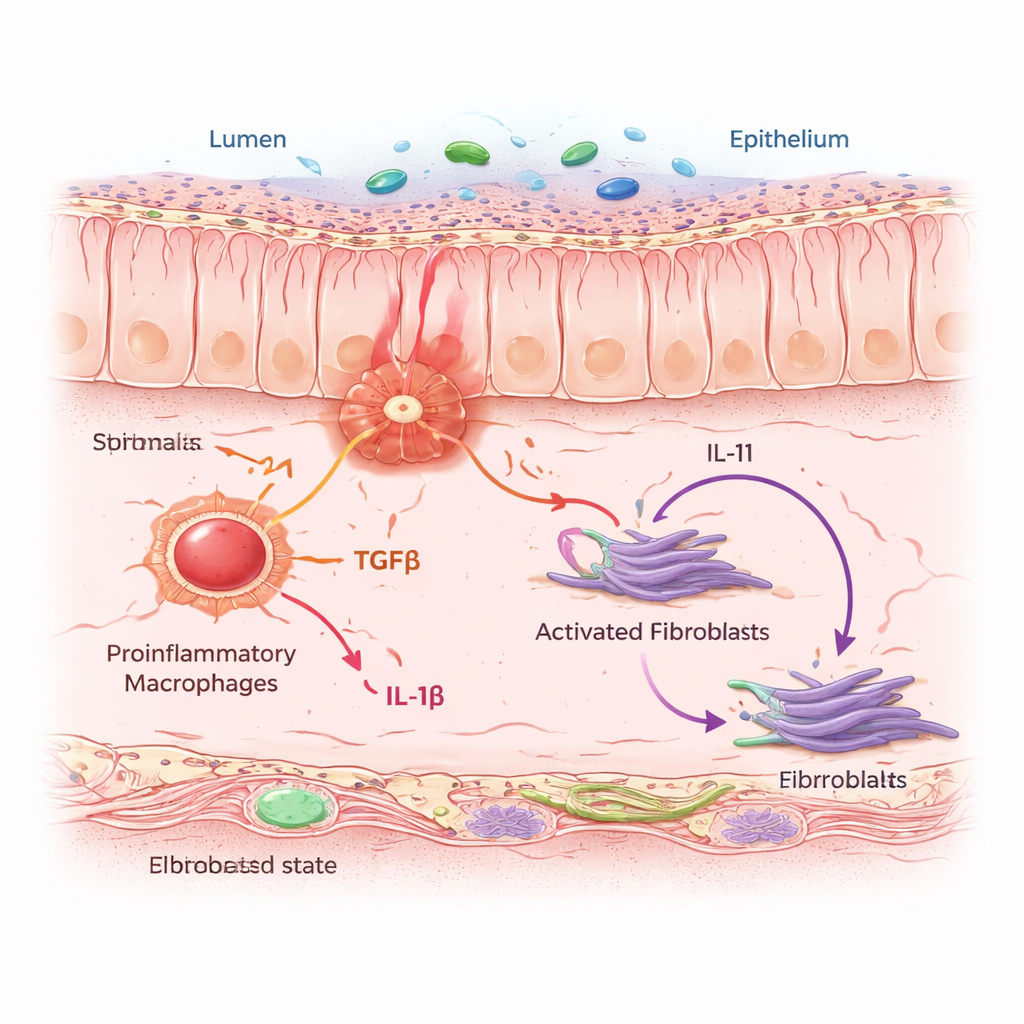
Макрофаги шепчут, фибробласты рубцуются
IAF не действовали в одиночку. Они образовывали «окрестности», плотные прoвоспалительными макрофагами — иммунными клетками, чувствующими опасность и выделяющими сигналы тревоги. С помощью вычислительных моделей и совместных культур клеток команда показала, что когда макрофаги переходят в воспалительное состояние, они секретируют два ключевых сигнальных белка: TGFβ и IL-1β. Поблизости фибробласты «слышат» эти сигналы через специфические рецепторы. Когда оба сигнала поступают одновременно, фибробласты переключаются в состояние IAF и начинают вырабатывать IL-11 — цитокин, ранее связанный с продвижением фиброза, — а также коллаген и другие матричные белки, утолщающие и уплотняющие стенку кишечника. У мышей, подвергнутых режиму хронического колита, блокирование IL-11 или его селективное удаление в фибробластах снижало накопление коллагена, не предотвращая при этом начальное воспаление, что показывает: IL-11 является ключевым драйвером фиброзной фазы.
GLIS3: главный переключатель в фибротических фибробластах
Чтобы перейти от корреляций к механизмам, авторы использовали мощные геномные CRISPR-инструменты. Они сконструировали человеческие фибробласты так, чтобы продукция IL-11 отслеживалась при помощи флуоресцентного маркера, а затем провели параллельные скрининги, в которых либо выключали, либо активировали гены по всему геному. Сортируя клетки, производившие необычно высокие или низкие уровни IL-11 после стимуляции TGFβ и IL-1β, они выявили гены, контролирующие эту реакцию. Среди множества сигнальных компонентов в качестве ключевого регулятора выделился один транскрипционный фактор — GLIS3. При дезактивации GLIS3 фибробласты вырабатывали значительно меньше IL-11; при его усилении уровень IL-11 резко возрастал. Дополнительные эксперименты показали, что GLIS3 перемещается в ядро фибробласта в ответ на сигналы макрофагов, непосредственно связывается с участками ДНК рядом с геном IL11 и другими генами и активирует широкую программу воспалительных и фибротических генов, включая коллагены и факторы, привлекающие дополнительные иммунные клетки. 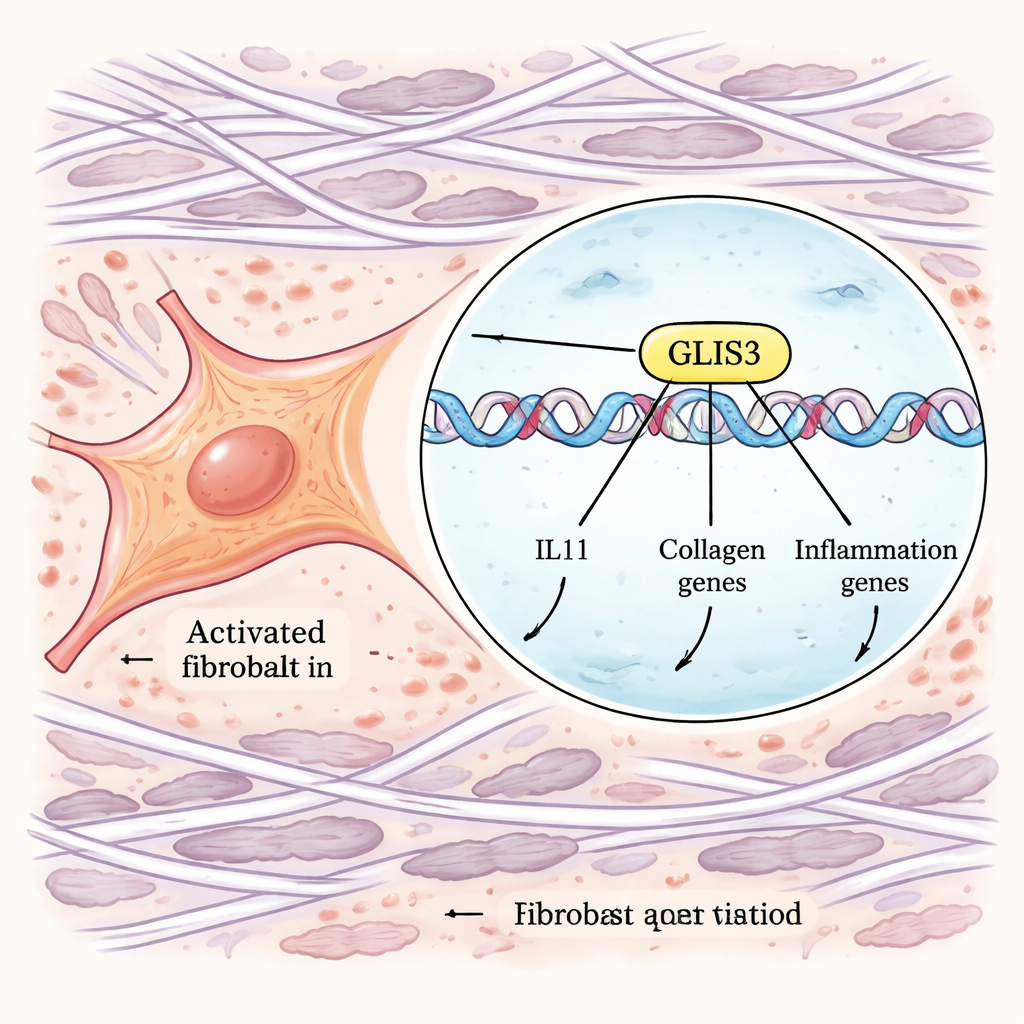
От моделей на мышах до тяжести заболевания у пациентов
Затем команда проверила, имеет ли значение программа, управляемая GLIS3, в живых организмах. У мышей создали линию, в которой GLIS3 можно было удалить только из фибробластов. При подвержении этих животных хроническому колиту они развивали меньше кишечного рубцевания, имели более низкие уровни коллагена и экспрессии фибротических генов и демонстрировали уменьшенное воспаление по сравнению с нормальными мышами. Пространственное картирование подтвердило, что у мышей, лишённых GLIS3, было меньше фибробластов, продуцирующих IL-11, и меньше рядом активированных макрофагов и нейтрофилов, что указывает на то, что нарушение GLIS3 ослабляет всю воспалительно-фибротическую цепочку. Обратившись к большой когорте детей с язвенным колитом, авторы выделили «подпись» GLIS3 из 50 генов и обнаружили, что её активность в биоптатах толстой кишки тесно коррелирует с тяжестью заболевания и числом IAF и активированных макрофагов, прямо связывая этот путь с исходами у пациентов.
Разорвать цикл воспаления и рубцевания
Для неспециалистов главный вывод таков: работа выявляет самоподдерживающийся цикл — воспалительные макрофаги заставляют фибробласты превращаться в рубцеобразующие IAF; эти IAF под контролем GLIS3 выделяют IL-11, коллаген и другие факторы, перестраивающие ткань и привлекающие больше воспалительных клеток. Стандартные лекарства, которые широко подавляют иммунную систему, могут не полностью разрывать этот цикл, что помогает объяснить, почему у многих пациентов со временем не удаётся достичь устойчивого ответа на имеющиеся терапии. Идентификация GLIS3 и состояния фибробластов, продуцирующих IL-11, как центральных узлов в цепи воспаление—фиброз указывает на более целевые стратегии — направленные на фибробласты, а не только на иммунные клетки — которые в будущем могли бы предотвратить или обратить рубцевание при воспалительных заболеваниях кишечника и, возможно, при других хронических воспалительных состояниях.
Цитирование: Pokatayev, V., Jaiswal, A., Shih, A.R. et al. Bidirectional CRISPR screens decode a GLIS3-dependent fibrotic cell circuit. Nature 650, 997–1006 (2026). https://doi.org/10.1038/s41586-025-09907-x
Ключевые слова: воспалительное заболевание кишечника, кишечный фиброз, фибробласты, макрофаги, GLIS3