Clear Sky Science · ru
Графовое разложение кластеров атомов для базовых межатомных потенциалов машинного обучения
Обучая компьютеры «чувствовать» атомы
Создание новых материалов для батарей, самолётов или термоядерных реакторов часто сводится к простому вопросу: как атомы толкают и тянут друг друга? Точное вычисление этих сил настолько затратно, что для одного материала на сверхкомпьютере могут уйти дни. В этой статье представлена новая семья моделей машинного обучения, названная GRACE, которая выступает как универсальный «калькулятор» для межатомных сил по большей части периодической таблицы. Их цель — сделать точные моделирования сложных материалов скорее рутинной задачей, а не героическим подвигом.
Одна модель для многих материалов
Большинство существующих силовых полей машинного обучения — это специализированные инструменты: они очень хорошо работают для нескольких элементов или соединений, но требуют полной перестройки при добавлении новых элементов. GRACE идёт иным путём. Она с самого начала разработана как базовая модель, способная охватить 89 химических элементов и огромное разнообразие атомных структур с единым набором правил. Для этого авторы опираются на математический аппарат, называемый разложением кластеров атомов, и расширяют его до графоподобных структур, что позволяет модели описывать как локальные окружения атомов, так и более протяжённые паттерны в единой форме. Вместо жесткого кодирования всех возможных взаимодействий GRACE обучается компактным «встраиваниям», которые фиксируют сходства между элементами, так что знания об одном материале помогают описывать другой.
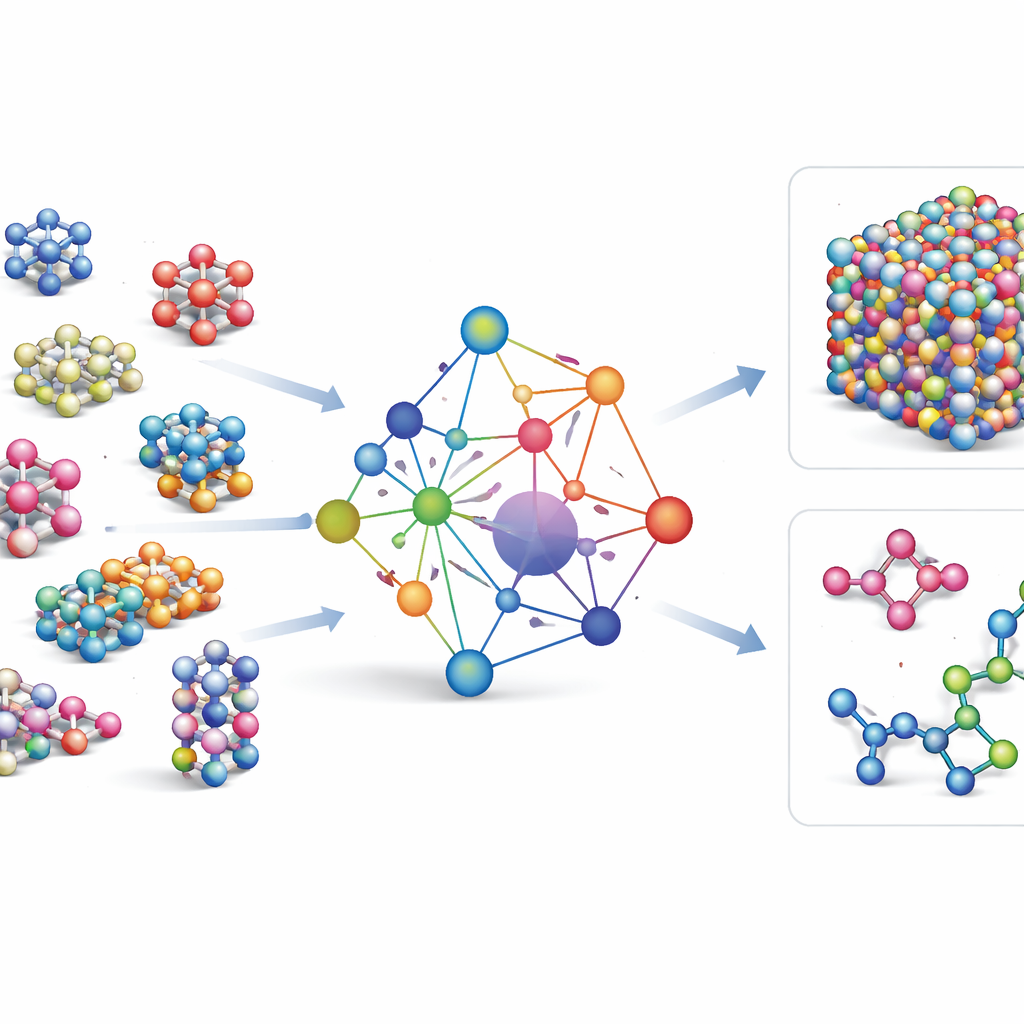
Обучение на море атомных данных
Чтобы научить GRACE, как ведут себя атомы, авторы собрали одни из крупнейших публичных баз данных квантовомеханических расчётов. Ядро составляют данные коллекции OMat24, которая включает около 110 миллионов симуляций неорганических материалов, дополненные двумя другими наборами, отслеживающими релаксацию и эволюцию структур. В совокупности эти наборы покрывают кристаллы рядом с равновесием, сжатые и искажённые структуры, высокотемпературные жидкости и многое другое по тому же широкому набору элементов. Модели GRACE выпускаются нескольких размеров — от простых однослойных версий, которые смотрят лишь на локальное окружение атома, до более глубоких двухслойных, которые эффективно передают «сообщения» между соседними регионами. Начальное обучение нацелено на сбалансированное предсказание энергий, сил и внутренних напряжений; последующая донастройка согласует модели с широко используемыми эталонными базами данных в материаловедении.
Проверка модели в деле
Универсальная модель полезна лишь в том случае, если она стабильно работает в разных задачах. Поэтому авторы подвергли GRACE требовательной тестовой программе, которая имитирует реальные сценарии использования атомистических симуляций учёными. На общем эталоне для поиска устойчивых кристаллических структур GRACE последовательно находится на «фронте Парето»: при заданной точности она быстрее конкурентов, а при заданной скорости — точнее. Аналогичные преимущества проявляются при предсказании теплопроводности, свойства, чувствительного к малейшим изменениям в движении атомов. GRACE также показывает хорошие результаты для упругих свойств, поверхностных энергий, энергий межзёренных границ и энергий образования точечных дефектов во многих чистых металлах — все эти тесты исследуют реакцию материалов на растяжение, резание или локальные повреждения. Долгий прогон молекулярной динамики горячей расплавленной соли показывает, что модель остаётся численно стабильной в течение наносекунд, воспроизводя детальные структурные шаблоны и скорости диффузии атомов.
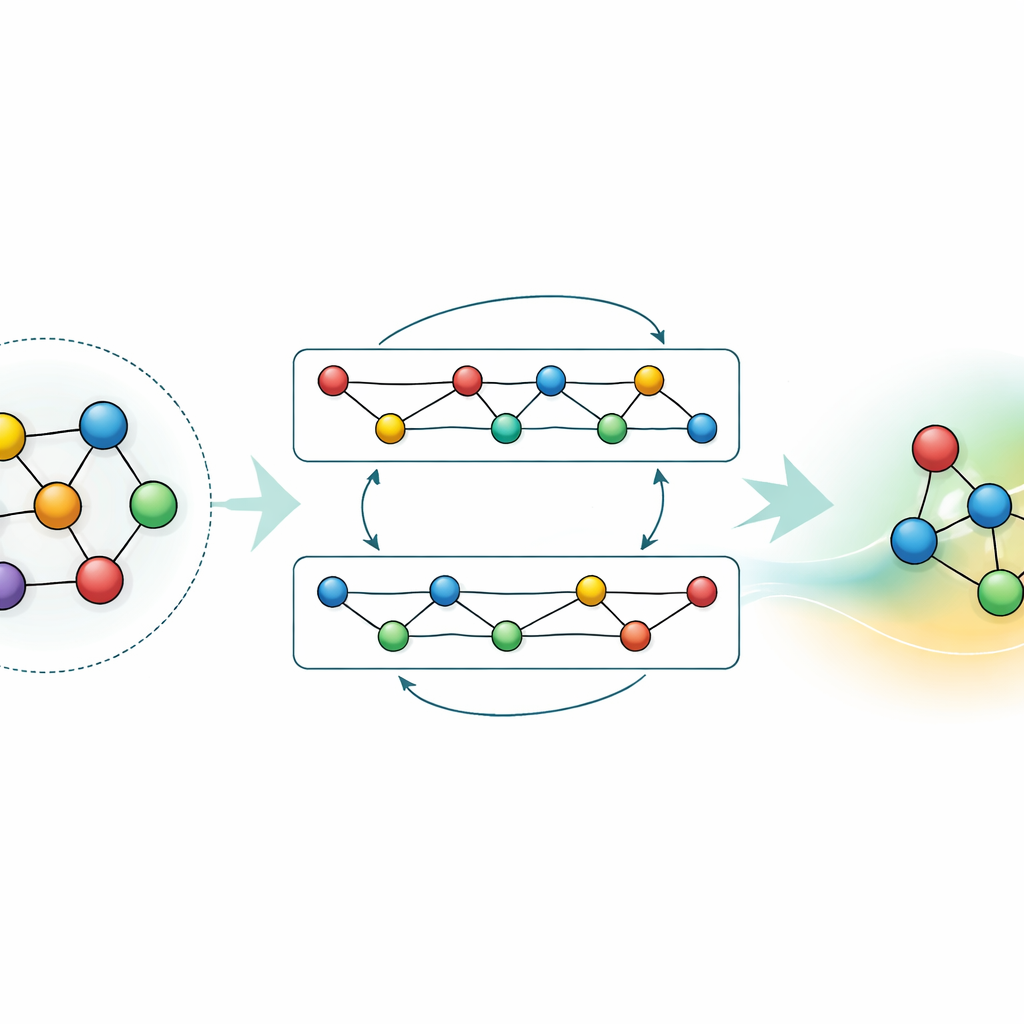
Адаптация и сжатие знаний
Несмотря на мощь универсальной модели, во многих приложениях требуется либо более высокая точность для конкретного материала, либо более быстрые расчёты на скромном железе. Авторы демонстрируют две стратегии, позволяющие этого добиться, не выбрасывая уже накопленные знания GRACE. Во‑первых, они дообучают базовую модель на узконаправленных наборах данных, например на алюминиево‑литиевых сплавах или на детальных путях горения водорода. Для сплавов даже умеренное дополнительное количество данных значительно улучшает предсказания, превосходя модели, обученные с нуля на тех же данных. Для задач горения наивная донастройка обычно приводила бы к «забыванию» остального материала; путём аккуратной фиксации частей сети и обновления лишь выбранных параметров авторы ограничивают катастрофическое забывание, одновременно повышая точность для новой химии. Во‑вторых, они показывают, как дистиллировать большую модель в гораздо более простого «студента», который имитирует учителя на ключевых системах. Эта дистиллированная версия работает примерно в семьдесят раз быстрее на CPU, сохраняя при этом большую часть точности, особенно если её обучать на смеси сложных сплавов и более простых эталонных структур, размеченных исходной GRACE.
Что это значит для будущего проектирования материалов
Работа позиционирует GRACE как гибкую основу для следующего поколения атомистического моделирования. Вместо разработки нового потенциала для каждого материала или свойства исследователи могут начать с универсальной модели GRACE и затем дообучить или дистиллировать её под свои нужды, экономя огромные объёмы компьютерного времени и экспертного труда. Бенчмарки показывают, что такой подход не просто сопоставим с существующими инструментами; во многих случаях он превосходит их по скорости и надёжности, особенно для сложных свойств, таких как тепловой транспорт. Для неспециалистов ключевое сообщение в том, что одна, хорошо продуманная модель машинного обучения теперь может выступать в роли широко доверенного «движка» для виртуальных экспериментов по большей части периодической таблицы, ускоряя поиск лучших батарей, катализаторов, конструкционных сплавов и энергетических материалов.
Цитирование: Lysogorskiy, Y., Bochkarev, A. & Drautz, R. Graph atomic cluster expansion for foundational machine learning interatomic potentials. npj Comput Mater 12, 114 (2026). https://doi.org/10.1038/s41524-026-01979-1
Ключевые слова: межатомные потенциалы машинного обучения, моделирование материалов, атомные симуляции, базовые модели, графовое разложение кластеров атомов