Clear Sky Science · ru
Патогенная мутация Tau вызывает дисфункцию аутофагии‑лизосом, что ограничивает деградацию Tau в модели фронтотемпоральной деменции
Когда уборочные бригады мозга отстают
Почему у некоторых людей появляются разрушительные нарушения памяти и поведения за десятилетия до старости? В этом исследовании вопрос рассматривают через призму одного белка мозга — Tau — и крошечных клеточных «перерабатывающих центров», которые обычно следят за его уровнем. Наблюдая за живыми человеческими нейронами под сверхточными микроскопами, исследователи показывают, как вызывающая болезнь мутация в Tau забивает систему утилизации клеток и как активация этой системы с помощью малой молекулы может помочь убрать накопления. Эти выводы могут указать путь к новым стратегиям лечения некоторых форм деменции.
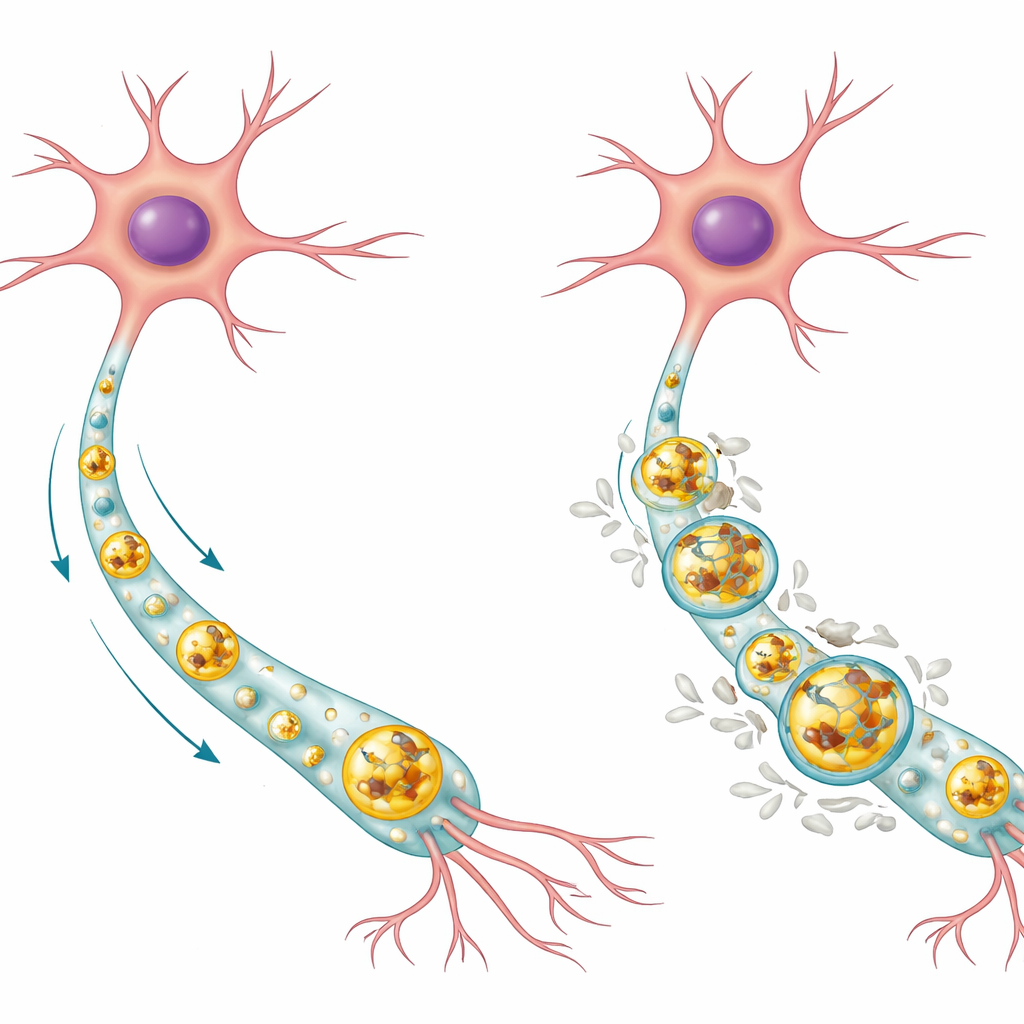
Как нейроны обычно выносят мусор
Нейроны — это долгоживущие клетки, которые не могут просто делиться, чтобы разбавить повреждённые компоненты, поэтому они сильно зависят от внутренних систем очистки. Один из ключевых путей — это путь аутофагии‑лизосом. В этом процессе нежелательные белки и изношенные части клетки заворачиваются в мембранные пузырьки — аутофагосомы, которые затем сливаются с наполненными ферментами компартментами — лизосомами, где груз расщепляется и перерабатывается. В здоровых человеческих нейронах авторы обнаружили, что нормальный белок Tau склонен накапливаться в кислой центральной части лизосом, где он может быть деградирован, в то время как фосфорилированная форма Tau (химическая модификация, связанная с болезнью) располагается преимущественно на внешней мембране лизосомы. Большинство лизосом в здоровых клетках вообще не содержали Tau, что указывает на то, что эта система обычно поддерживает низкий и контролируемый уровень Tau.
Что идёт не так при генетической форме деменции
Команда сосредоточилась на мутации в гене MAPT, называемой p.R406W, которая вызывает наследственную форму фронтотемпоральной деменции и может имитировать ухудшение памяти, похожее на болезнь Альцгеймера. С помощью технологий стволовых клеток они перепрограммировали кожные клетки пациентов в индуцированные плюрипотентные стволовые клетки, а затем в большое количество человеческих нейронов, которые либо несли эту мутацию, либо были генетически отредактированы обратно к норме. В мутантных нейронах общие уровни Tau и фосфорилированного Tau были значительно выше — не потому, что клетки синтезировали больше Tau, а потому, что они хуже его удаляли. Суперразрешающая визуализация показала, что почти все лизосомы в мутантных клетках были забиты Tau и особенно фосфорилированным Tau, покрывающим мембрану лизосомы. Это накопление сигнализировало о том, что главный путь утилизации белков в клетке был заблокирован.
Забитые перерабатывающие центры и замедленное движение
При более тщательном изучении механики переработки исследователи обнаружили, что лизосомы в мутантных нейронах были более многочисленными, крупнее и, как правило, располагались дальше от тела клетки. Живое изображение с флуоресцентными красителями показало, что эти лизосомы двигались медленнее и преодолевали меньшие расстояния вдоль нервных отростков, хотя сами микротрубочки, служащие путями, выглядели нормально. В мутантных нейронах также было больше аутофагосом, больше белка‑адаптера p62 и дополнительные липидные капли — признаки того, что материалы помечаются для утилизации, но не полностью расщепляются. С помощью pH‑чувствительного репортера они обнаружили, что аутофагосомы в мутантных клетках часто не сливаются должным образом с лизосомами, что приводит к накоплению «наполовину готовых» перерабатывающих везикул и широкому нарушению очистки клетки, затрагивающему не только Tau, но и другие грузы.
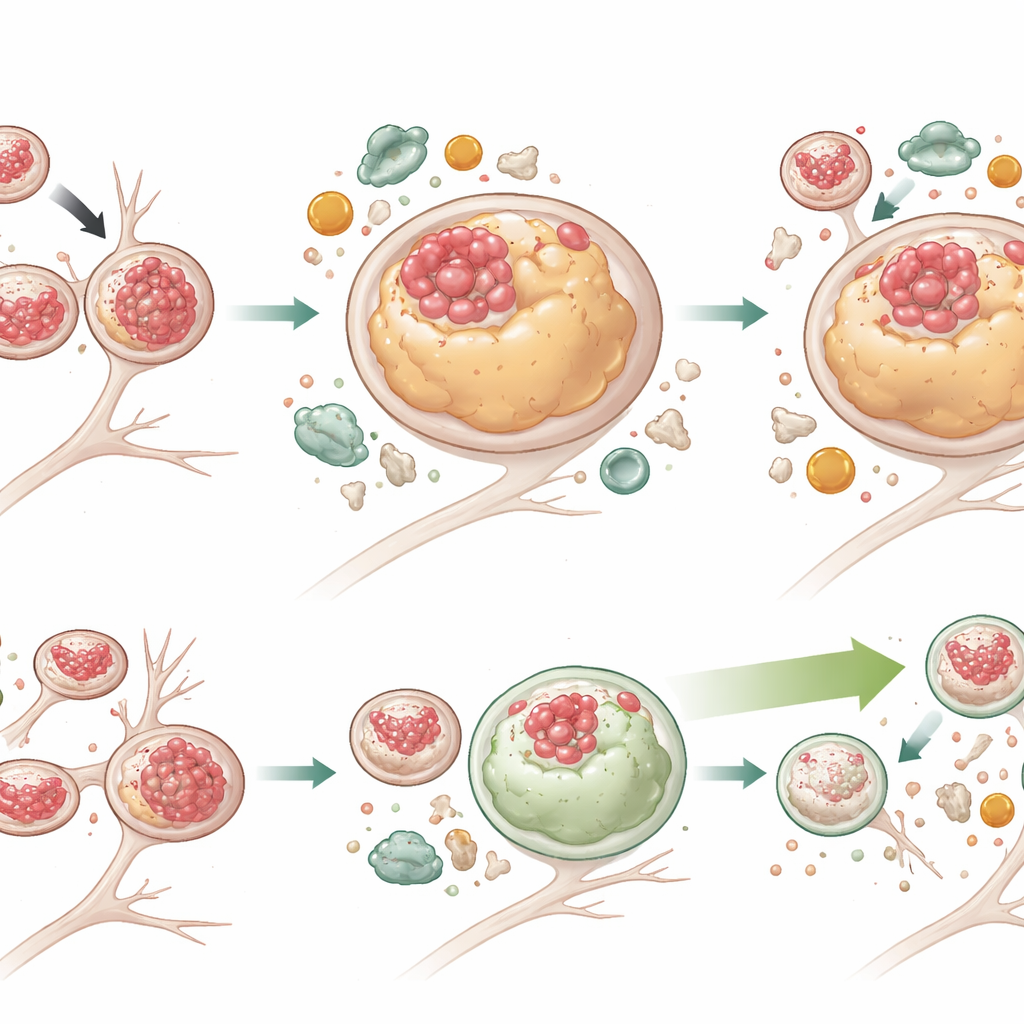
Усиление очистки клетки без устранения пробки в движении
Чтобы проверить, может ли усиление аутофагии преодолеть эти проблемы, команда обработала нейроны G2‑567, малой молекулой, ранее показанной как стимулятор системы аутофагии‑лизосом. После двух недель лечения в мутантных нейронах существенно снизились уровни как общего Tau, так и фосфорилированного Tau, и многие лизосомы вновь оказались свободны от Tau. Лизосомы также уменьшились в размере, приблизившись к норме. Маркеры активной аутофагии увеличились, тогда как p62 — индикатор застрявшей деградации — уменьшился в мутантных клетках, демонстрируя более эффективное расщепление груза. Интересно, что G2‑567 не устранял все дефекты: лизосомы в мутантных нейронах по‑прежнему обычно располагались дальше от тела клетки и двигались вяло, а уровень адаптерного белка (JIP3), связанного с транспортом лизосом, оставался повышенным. Это говорит о том, что функции перемещения и деградации лизосом могут быть частично разграничены, и что улучшение только процессов расщепления может быть достаточным, чтобы уменьшить токсичное накопление Tau.
Что это значит для будущих методов лечения деменции
Для неспециалиста главный вывод таков: в этой генетической модели фронтотемпоральной деменции проблема заключается не просто в том, что Tau становится аномальным; она в том, что система переработки нейрона не успевает за нагрузкой. Мутация p.R406W в Tau напрямую нарушает несколько шагов пути аутофагии‑лизосом, вызывая накопление Tau — особенно его фосфорилированной формы — на поверхности и внутри лизосом вместе с другими недеградированными материалами. Фармакологическое стимулирование клеточных механизмов очистки позволило исследователям снизить уровни Tau и нормализовать размер лизосом, хотя нарушения транспорта оставались. Эти результаты укрепляют идею о том, что препараты, безопасно усиливающие аутофагию и функцию лизосом, могут помочь восстановить белковый гомеостаз при тау‑связанных деменциях и, возможно, при более распространённых состояниях, таких как болезнь Альцгеймера.
Цитирование: Mirfakhar, F.S., Marsh, J.A., Sato, C. et al. A pathogenic Tau mutation drives autophagy-lysosome dysfunction that limits Tau degradation in a model of frontotemporal dementia. Nat Commun 17, 2699 (2026). https://doi.org/10.1038/s41467-026-70473-5
Ключевые слова: белок тау, аутофагия, дисфункция лизосом, фронтотемпоральная деменция, нейродегенерация