Clear Sky Science · ru
Эффективная выборка масштабных переходных путей и промежуточных конформаций в субмезоскопических белковых комплексах
Наблюдение за белками в движении
Многие молекулы, которые поддерживают нашу жизнь, ведут себя не как жёсткие кирпичики Lego, а скорее как крошечные машины, постоянно меняющие форму. Эти движения питают процессы, такие как выработка энергии, ремонт ДНК и проникновение вирусов в клетки. Эксперименты, например крио-электронная микроскопия, теперь могут «заморозить» некоторые из этих форм, но не мимолётные шаги между ними. В этой статье представлена eBDIMS2 — новый компьютерный метод, который может «дозаполнить отсутствующие кадры» движения белка даже для огромных молекулярных машин, которые ранее были слишком большими и сложными для моделирования на обычном компьютере.
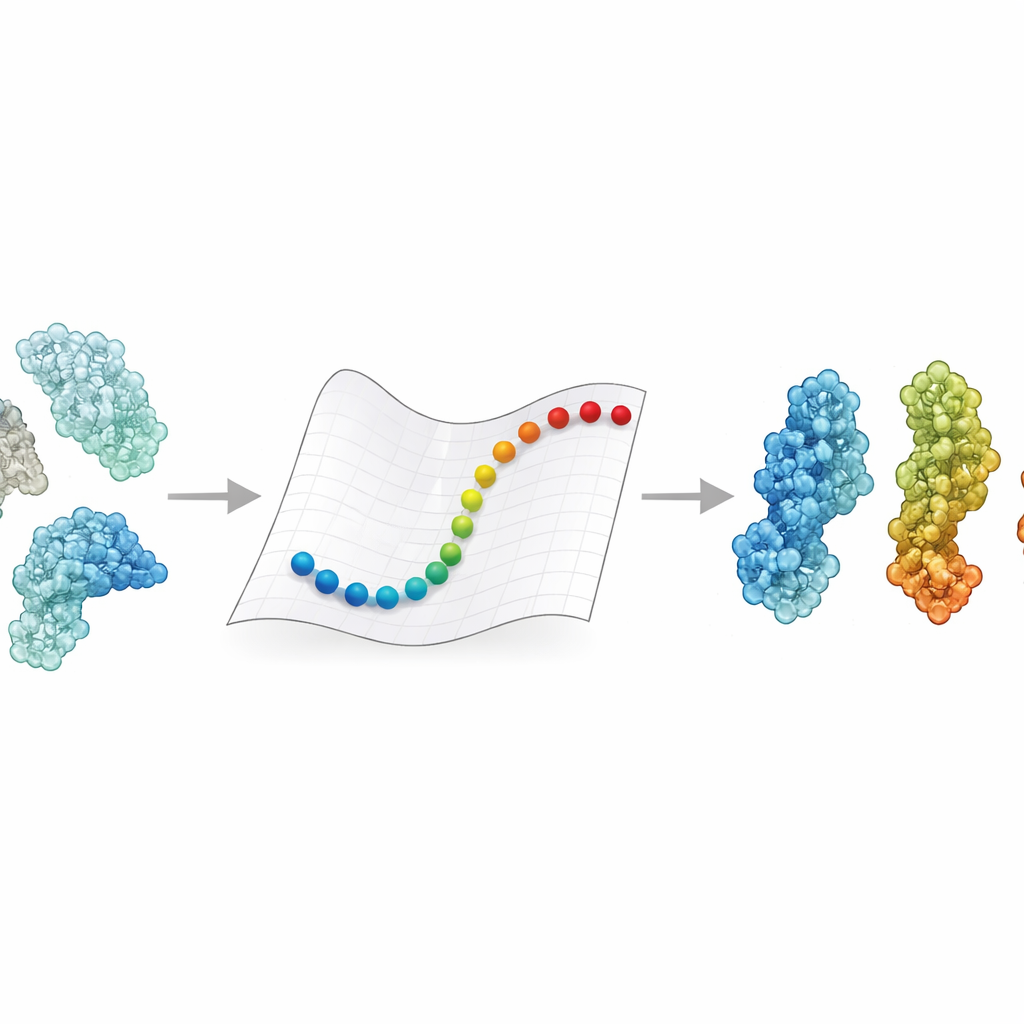
Почему изменение формы белка важно
Белки редко остаются застывшими в одной позе. Они открываются и закрываются, скручиваются и сгибаются в ответ на сигналы — изменение напряжения, pH или связывание партнёра. Эти сдвиги могут означать разницу между активностью и неактивностью фермента или между захватом вируса рецептором и его утратой. Эксперименты дают детальные снимки нескольких ключевых форм, а молекулярная динамика принципиально может связать их, прослеживая каждый атом во времени. Но отслеживание таких движений для огромных комплексов, которые теперь видны с помощью крио-ЭМ и часто весят сотни тысяч — миллионы дальтон, обычно требует суперкомпьютеров и недель вычислений. В результате для многих медицински значимых гигантов мы до сих пор не знаем, как одно состояние переходит в другое.
Быстрый путь через конформационные ландшафты
eBDIMS2 берёт короткий путь, упрощая представление белков и расчёт их движений. Вместо того чтобы отслеживать каждый атом, метод рассматривает каждую аминокислоту как одну точку, соединённую пружинами в упругой сети. Эти пружины фиксируют, какие части белка склонны двигаться вместе. Затем метод использует броуновскую динамику — математические правила, имитирующие качание в жидкости — чтобы подтолкнуть структуру от одного экспериментально известного состояния к другому. Важно, что eBDIMS2 учитывает лишь те взаимодействия, которые действительно имеют значение, применяя отсечки по расстоянию и параллельные вычисления для снижения затрат. Это улучшает масштабирование программы примерно с квадратичного до почти линейного по размеру белка. На практике это означает, что переходы для огромных сборок, приближающихся к двум миллионам дальтон, можно исследовать за часы на настольном компьютере, вместо того чтобы они были практически недоступны.
Сверка путей с реальными белками
Чтобы проверить, имеют ли эти быстрые пути биологический смысл, авторы собрали ансамбли из 47 крупных белков и ещё 15 комплексов, всего сотни структур, в основном решённых методом крио-ЭМ. Они использовали анализ главных компонент — статистический инструмент, который выделяет доминирующие способы движения каждого белка — чтобы организовать эти структуры в конформационные ландшафты, такие как «открыто», «закрыто», «активно» или «неактивно». Затем eBDIMS2 попросили связать пары конечных состояний на этом ландшафте. Получившиеся пути проецировали обратно на те же низкоразмерные карты, чтобы посмотреть, проходят ли они гладкими маршрутами рядом с экспериментально наблюдаемыми промежуточными состояниями. В более чем в 30% систем смоделированные маршруты проходили близко — в пределах нескольких ангстрем — к промежуточным структурам, которые не были предоставлены как входные данные. Для сложных случаев, таких как фермент репарации ДНК DNA-PKcs или шиповидный белок коронавируса, упрощённые конформационные пути также хорошо совпадали с гораздо более затратными атомными симуляциями, включая целенаправленную молекулярную динамику и продвинутые методы повышения выборки.
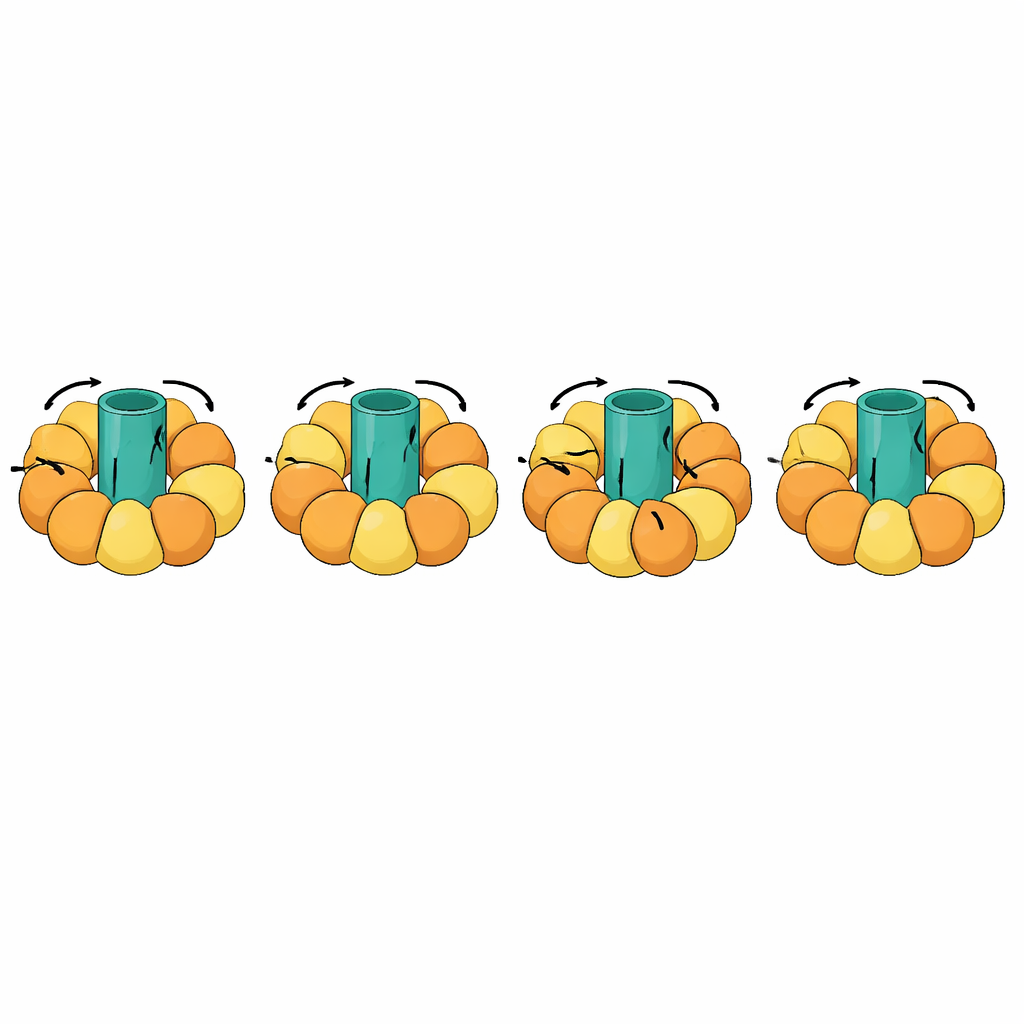
Прослеживание гигантских молекулярных машин
Один из самых впечатляющих тестов касался роторовых машин, таких как АТФ-синтазы, которые производят клеточную «энергетическую валюту», связывая вращающийся ротор в мембране с открыванием и закрыванием движений в окружающих субъединицах. Эти переходы исключительно сложны: части молекулы должны оставаться жёсткими и вращаться как единое целое, в то время как другие фрагменты гибко работают в хореографическом цикле. eBDIMS2 вводит специальную обработку для таких квазижёстких частей и для неполных экспериментальных моделей с пропущенными сегментами, что часто встречается в крио-ЭМ. Благодаря этим возможностям он может моделировать полные циклы вращения АТФ-синтазы и других массивных комплексов, таких как молекулярные шапероны, рецепторы и вирусные сборки. При этом сгенерированные промежуточные структуры избегают серьёзных искажений, которые дают некоторые конкурирующие методы, и их можно «очистить» до атомистических моделей, пригодных для расчётов дизайна лекарств или более длинных, детальных симуляций.
Что это значит для биологии и медицины
Исследование показывает, что eBDIMS2 способен надёжно набросать основные маршруты между известными формами белков для систем, недоступных традиционным симуляциям. Он не заменяет детальные атомные «фильмы» и не даёт точных энергий и временных масштабов, но предлагает быстрый, физически обоснованный способ картирования того, как могут двигаться большие молекулярные машины, используя только пару экспериментально определённых структур в качестве входа. По мере того как структурные базы данных пополняются множественными состояниями больших белковых ассамблей, связанными с раком, инфекциями и другими заболеваниями, этот подход даёт исследователям доступный инструмент для «связывания точек», предложения правдоподобных промежуточных состояний и указания, куда стоит направить последующие исследования с более высоким разрешением или целенаправленным дизайном препаратов.
Цитирование: Scaramozzino, D., Lee, B.H. & Orellana, L. Efficient sampling of large-scale transition pathways and intermediate conformations in sub-mesoscopic protein complexes. Nat Commun 17, 2202 (2026). https://doi.org/10.1038/s41467-026-69809-y
Ключевые слова: динамика белков, молекулярные моделирования, крио-ЭМ, конформационные пути, упрощённое моделирование