Clear Sky Science · ru
Активация IRF3 в кардиомиоцитах нарушает митохондриальную окислительную функцию через подавление PGC-1α и ведёт к сердечной недостаточности
Почему важны уставшее сердце и истощённые клетки
Сердечную недостаточность часто описывают как «износ» сердца, но за этой метафорой скрывается хроника воспаления и истощённых энергетических станций внутри кардиомиоцитов. В этом исследовании задаётся на первый взгляд простой, но значимый вопрос: существует ли единый молекулярный переключатель в клетках сердца, который связывает вредное воспаление и провал энергетического производства — и можно ли, изменив состояние этого переключателя, повлиять на ход сердечной недостаточности? Следуя этой нити, авторы выявляют ключевого участника и показывают, что мягкое усиление собственных энергетических программ сердца частично спасает сердца мышей от прогрессирования заболевания.
Молекулярный выключатель в больных человеческих сердцах
Исследователи сосредоточились на белке IRF3, наиболее известном своей ролью в ответе клеток на вирусные инфекции. Они изучали ткани людей с ишемической кардиомиопатией, распространённой формой сердечной недостаточности, возникающей после инфарктов из‑за снижения кровоснабжения. В этих поражённых сердцах IRF3 был не просто обнаружен — он был химически активирован в определённых участках, что указывает на его участие в включении программ генов. Одновременно аппаратура митохондрий, преобразующая топливо в энергию через окислительное фосфорилирование, была заметно ослаблена. Схожая картина наблюдалась и в мышиных моделях инфаркта: при перевязке коронарной артерии IRF3 в кардиомиоцитах сильно активировался, и гены, контролируемые IRF3, включались. Даже фрагменты митохондриальной ДНК — высвобождённые из повреждённых митохондрий и выступающие как внутренние «сигналы опасности» — были достаточны, чтобы включить IRF3 в изолированных кардиомиоцитах.
Выключение IRF3 защищает сердце
Чтобы проверить, усугубляет ли активность IRF3 заболевание, команда создала мышей, у которых IRF3 можно было удалить исключительно из кардиомиоцитов, не затрагивая иммунные и поддерживающие клетки. После индуцированного инфаркта у этих мышей была лучшая сократительная функция и меньше рубцевания по сравнению с обычными мышами, несмотря на одинаковую исходную травму. В выращенных в чашках сердечных клетках подавление IRF3 снижало уровень провоспалительных генов, не нарушая при этом работу родственных белков. В совокупности эти данные свидетельствуют: IRF3 внутри самой сердечной клетки — не пассивный наблюдатель; он усиливает воспаление и структурные повреждения после ишемии и способствует переходу к сердечной недостаточности.
Когда IRF3 застревает «включённым», система питания рушится
Авторы провели обратный эксперимент: они создали мышей, в которых IRF3 в кардиомиоцитах можно было перевести в постоянно активное состояние с помощью генетического «фосфомиметического» приёма. Даже без внешнего триггера эти животные быстро развивали тяжёлую сердечную дисфункцию, высокий уровень воспалительных молекул в крови и признаки клеточного повреждения. Глубокий анализ ткани сердца показал, что при хронической активности IRF3 подавляется главный координатор энергетического метаболизма — PGC-1α. Этот белок обычно поддерживает здоровые митохондрии, эффективное использование жиров и сбалансированную клеточную энергию. При понижении PGC-1α падали уровни множества митохондриальных белков, давал сбой электронно‑транспортная цепь, и менялись предпочтения сердца в выборе топлива: снижались карнитин и связанные с утилизацией жиров соединения, ухудшалось использование кетонов, а обращение с глюкозой было нарушено. Даже соотношение NAD⁺/NADH — важный показатель редокс‑баланса клетки — сдвигалось в неблагоприятную сторону.
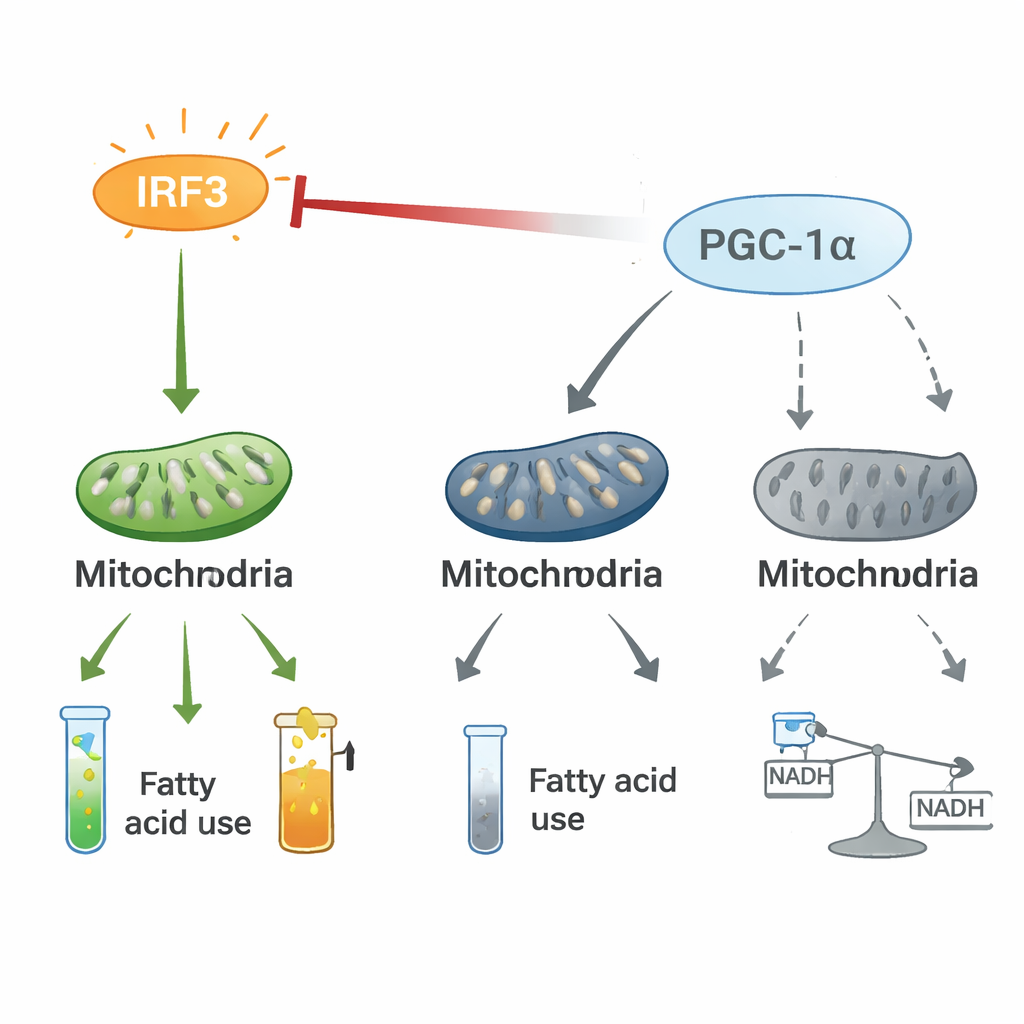
Перетягивание каната между воспалением и контролем энергии
Механистические эксперименты показали, что IRF3 и PGC-1α образуют двунаправленную регуляторную ось. В кардиомиоцитах активированный IRF3 физически взаимодействует с PGC-1α и ослабляет его способность включать гены жирового окисления. Подавление IRF3 повышает уровни и активность PGC-1α, тогда как усиление PGC-1α снижает экспрессию IRF3‑опосредованных провоспалительных генов и восстанавливает митохондриальные маркеры, даже при стрессах вроде низкого кислорода или бактериальных токсинов. Исследования со стабильными изотопами показали, что активация IRF3 перенаправляет углерод из обычного энергетического потока через цикл трикарбоновых кислот в альтернативный путь — пентозофосфатный — и нарушает плавное течение метаболитов. Это перетягивание каната между провоспалительным переключателем (IRF3) и энергетическим партнёром (PGC-1α) по‑видимому перестраивает метаболизм сердца в пользу воспаления и потери энергии.
Мягкая перезарядка «батарей» сердца
Наконец, команда спросила, сможет ли умеренное повышение PGC-1α противодействовать вреду, вызываемому IRF3. Они использовали нацеленный на сердце генотерапевтический вектор, чтобы умеренно — но не чрезмерно — поднять уровни PGC-1α в тех же мышах с гиперактивным IRF3. Это скромное повышение улучшило сократительную функцию, увеличило содержание митохондриальных белков, усилило экспрессию генов жирового окисления и метаболизма NAD, а также снизило активность воспалительных и фибротических генов. В клеточных экспериментах совместная экспрессия PGC-1α с активным IRF3 восстановила более здоровый баланс NAD⁺/NADH и вернула использование топлива в сторону жиров. Для непрофессионального читателя это означает, что аккуратная перезарядка системы управления «батареями» сердца может частично компенсировать вред хронического воспалительного переключателя, застрявшего в положении «включено».
Что это значит для будущего лечения сердечной недостаточности
Эта работа выделяет IRF3 как центральное звено между воспалением и энергетическим коллапсом внутри кардиомиоцитов. Вместо того чтобы рассматривать воспаление и метаболизм как разрозненные задачи при сердечной недостаточности, исследование предлагает их взаимосвязанность через ось IRF3–PGC-1α. Хотя выводы получены на моделях мышей и клеток, они создают основание для гипотезы, что будущие терапии могут либо уменьшать активность IRF3, либо укреплять PGC-1α и функцию митохондрий, чтобы замедлить или предотвратить сердечную недостаточность после инфаркта. Проще говоря, успокоение избыточной клеточной «сигнализации тревоги» и поддержка энергетических фабрик сердца могут оказаться мощной комбинированной стратегией, позволяющей ослабленным сердцам дольше биться эффективно.
Цитирование: Kumari, M., Evangelakos, I., Deshpande, A. et al. Activation of IRF3 in cardiomyocytes impairs mitochondrial oxidative function through PGC-1α inhibition and drives heart failure. Nat Commun 17, 2051 (2026). https://doi.org/10.1038/s41467-026-69792-4
Ключевые слова: сердечная недостаточность, воспаление, митохондрии, кардиомиоциты, PGC-1α