Clear Sky Science · ru
Прогнозирование химического сходства между тандемными масс-спектрами в разных режимах ионизации в метаболомике
Почему важно связывать химические точки
Каждый глоток кофе, вдох воздуха или приём лекарства оставляет в нашем организме крошечные химические следы. Современные приборы могут одновременно обнаруживать тысячи таких молекул, но превращение этих сигналов в биологическое понимание остаётся удивительно сложной задачей. В этом исследовании представлена MS2DeepScore 2.0 — инструмент машинного обучения, который помогает учёным увидеть, как эти молекулы связаны между собой, даже когда сигналы регистрируются очень по-разному. Это обещает более быстрые и полные интерпретации сложных химических смесей в медицине, питании и экологических исследованиях.
Два взгляда на одну и ту же молекулу
Масс-спектрометрия — это основная методика, которая взвешивает и разрушает молекулы, раскрывая их идентичность. В рутинных экспериментах учёные часто измеряют один и тот же образец в двух режимах: в режиме, благоприятствующем положительно заряженным ионов, и в режиме, благоприятствующем отрицательно заряженным. Каждый режим даёт свой характерный «штрих-код» фрагментов. Даже когда оба измерения получены от одной молекулы, получающиеся паттерны могут выглядеть настолько по-разному, что традиционные методы сравнения не срабатывают. В результате исследователи обычно анализируют два режима раздельно, строят два несвязанных отображения образца и рискуют упустить важные связи между веществами.
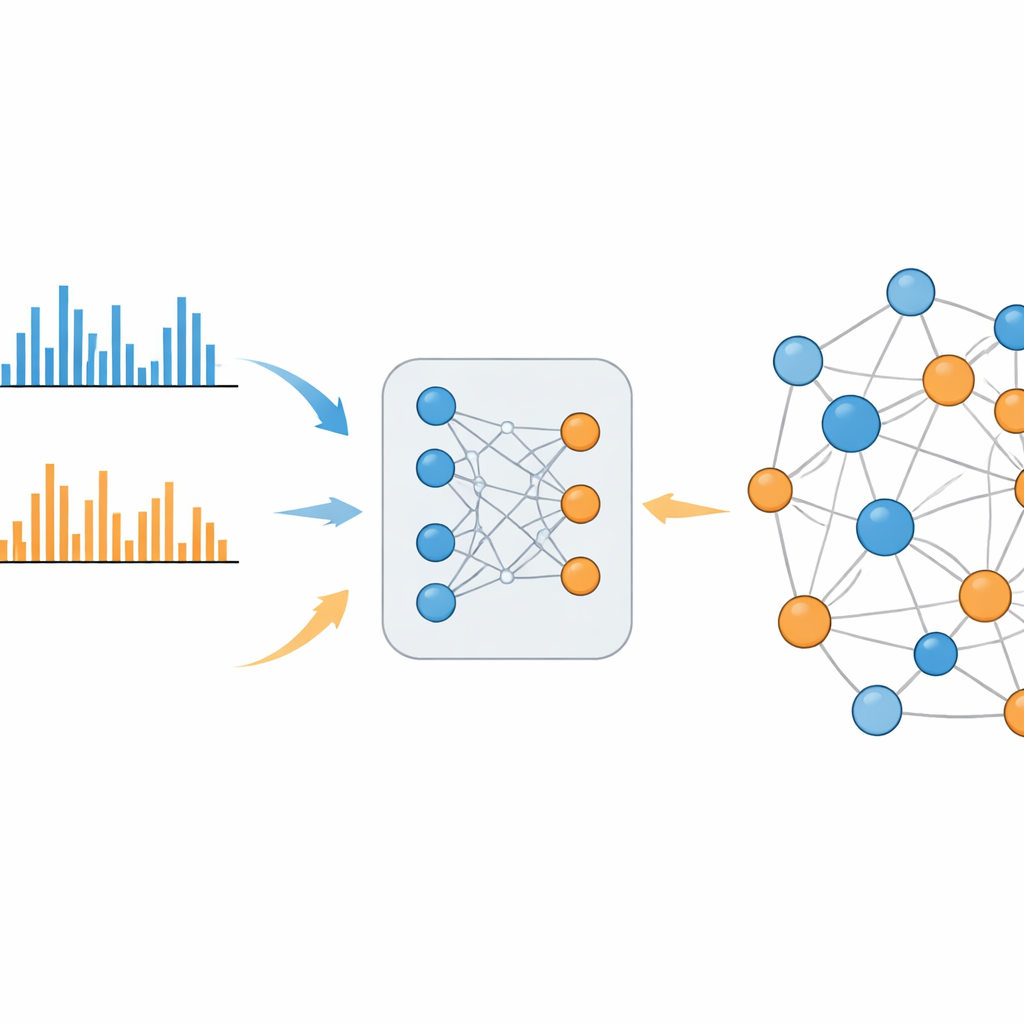
Обучаемая система, преодолевающая разрыв
MS2DeepScore 2.0 решает эту проблему, обучаясь определять химическое сходство напрямую на больших библиотеках известных спектров. Модель построена по схеме «близнецовых» нейронных сетей, которые преобразуют каждый паттерн фрагментации в вектор-отпечаток из 500 чисел, называемый embedding. Во время обучения система видит сотни тысяч примеров как из положительного, так и из отрицательного режимов, а также информацию о том, насколько реально схожи исходные молекулы. Она подстраивается так, чтобы спектры родственных молекул получали похожие embeddings, независимо от того, измерялись ли они в одном и том же режиме или в противоположных. Новая версия расширяет возможности предшественника, добавляя дополнительную информацию, такую как масса исходной молекулы и использованный режим ионизации, а также применяя тщательно сбалансированную схему выборки, чтобы редкие, но информативные химические связи не терялись на фоне обычных, малоинформативных совпадений.
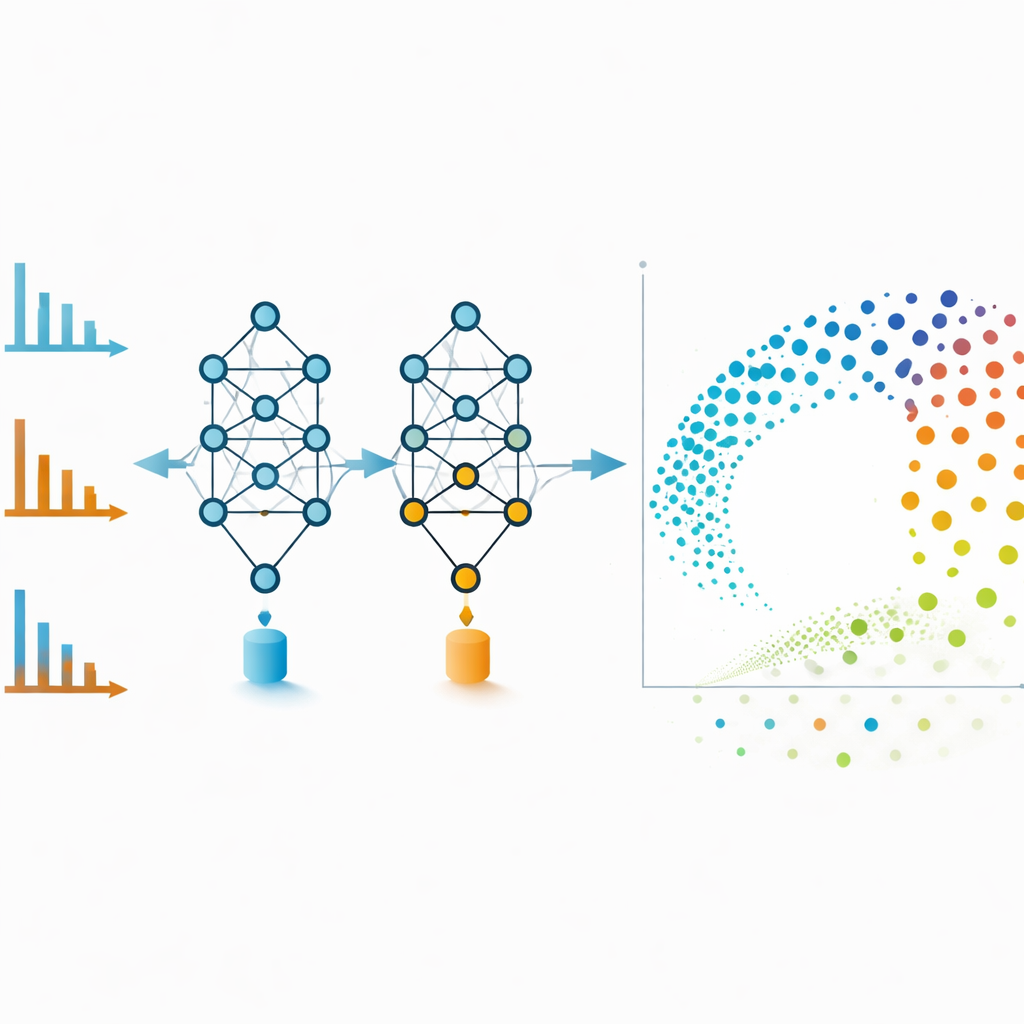
От разрозненных сигналов к единым картам
После обучения MS2DeepScore 2.0 может оценивать, насколько химически схожи любые два спектра, включая пары «плюс» против «минус». Авторы показывают, что эти предсказания хорошо коррелируют с устоявшимися мерами структурного сходства, не только внутри каждого режима, но и между режимами. Используя реальные данные из мочи человека, плазмы крови и дикорастущего съедобного растения, они строят «молекулярные сети», в которых каждый спектр — это узел, а сильное предсказанное сходство создаёт связь. В отличие от старых подходов, эти сети естественно смешивают данные из положительного и отрицательного режимов в единые, согласованные карты. Кластеры, отобранные экспертами, включают, например, группы молекул, связанных с кофеином в моче, которые связаны через режимы ионизации и соответствуют известным метаболическим путям.
Видеть химический ландшафт одним взглядом
Молекулярные сети мощны, но могут запутываться, если включать слишком много слабых связей. Чтобы избежать этого, авторы используют embeddings MS2DeepScore напрямую как координаты в двумерной раскладке, созданной с помощью техники UMAP. Каждая точка на этой карте представляет один спектр, а близкие точки соответствуют молекулам, которые модель считает химически похожими. Спектры положительного и отрицательного режимов одного и того же соединения, которые визуально выглядят очень по-разному, часто оказываются рядом в этом пространстве embedding-ов. Команда также обучает дополнительную модель, которая оценивает надёжность каждого embedding-а, помечая спектры, которые шумны, неполны или не похожи ни на что, что встречалось в обучении. Удаление таких низкокачественных точек повышает общую точность и делает визуализации более достоверными.
Продвинутые инструменты для повседневных лабораторий
Чтобы технология была доступна не только программистам, авторы интегрировали MS2DeepScore 2.0 в популярное свободно распространяемое программное обеспечение для масс-спектрометрии. Благодаря этой интеграции исследователи могут обнаруживать признаки, строить молекулярные сети, игнорирующие границы режимов ионизации, и изучать полученное химическое пространство через интерактивные панели. Код, обученные модели и примерные наборы данных открыто доступны, а систему можно дообучить или тонко настроить для специализированных классов соединений.
Что это значит для будущих открытий
Для неспециалистов главное сообщение таково: MS2DeepScore 2.0 помогает превратить фрагментированные и зависящие от режима измерения в единый, более понятный образ молекул, присутствующих в образце. Надёжно связывая сигналы, которые раньше жили в раздельных аналитических мирах, метод позволяет учёным использовать гораздо большие справочные библиотеки, полнее сравнивать образцы и сосредоточивать внимание на значимых кластерах родственных соединений. Такие сквозные связи данных, как ожидается, ускорят идентификацию биомаркеров, нутриентов, природных продуктов и загрязнителей, в конечном счёте углубляя наше понимание того, как химия влияет на здоровье и окружающую среду.
Цитирование: de Jonge, N.F., Chekmeneva, E., Schmid, R. et al. Cross ionization mode chemical similarity prediction between tandem mass spectra in metabolomics. Nat Commun 17, 2483 (2026). https://doi.org/10.1038/s41467-026-69083-y
Ключевые слова: метаболомика, масс-спектрометрия, машинное обучение, молекулярные сети, химическое сходство