Clear Sky Science · ru
Оценка технологий атласирования single-cell ATAC-seq с помощью моделирования от последовательности к функции
Чтение инструкции клетки
Каждая клетка в вашем организме считывает одну и ту же ДНК, но нейроны, мышечные и иммунные клетки ведут себя по‑разному. В этой статье рассматривается ключевая загадка такого разнообразия: как короткие участки ДНК, называемые энхансерами, действуют как переключатели, включая и выключая гены в определённых типах клеток. Авторы показывают, что новые, более дешёвые лабораторные технологии могут создавать массивные наборы данных, необходимые для обучения современных моделей глубокого обучения, которые читают последовательности ДНК и предсказывают, какие энхансеры активны в каких клетках, приближая нас к расшифровке регуляторной «грамматики» генома.
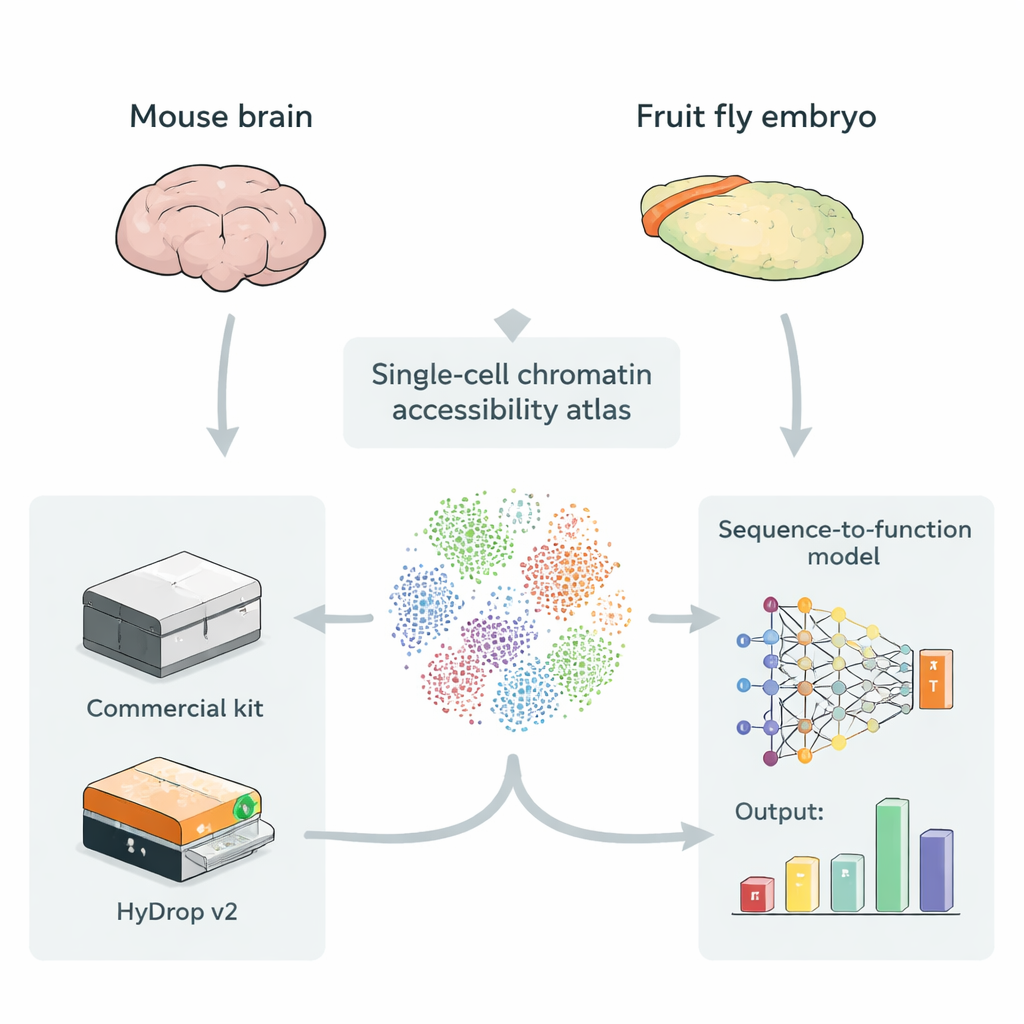
Создание карт открытой ДНК в одиночных клетках
Энхансеры обычно располагаются в участках ДНК, которые более открыты и доступны, что облегчает связывание регуляторных белков. Метод single‑cell ATAC‑seq измеряет, какие части генома открыты в тысячах и даже сотнях тысяч отдельных клеток одновременно, создавая «атлас» доступной ДНК по множеству типов клеток. Такие атласы идеальны для моделей глубокого обучения, которые берут на вход сырую последовательность ДНК и учатся предсказывать, насколько сильно каждый небольшой участок действует как энхансер в каждом типе клеток. До сих пор большинство таких атласов получали с помощью дорогих коммерческих приборов, поэтому возникал вопрос, могут ли недорогие открытые методы дать тренировочные данные равной ценности для этих моделей.
Открытый аналог коммерческих платформ
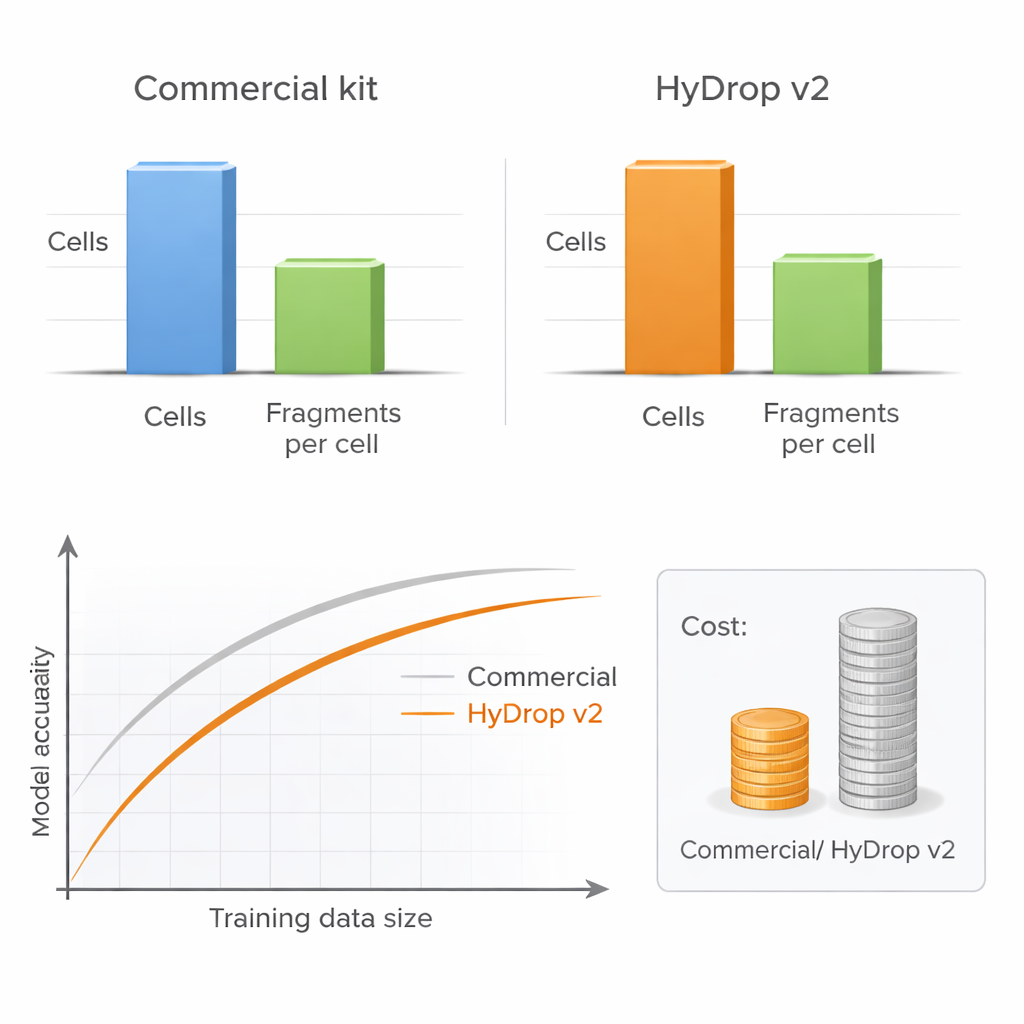
Авторы представляют HyDrop v2 — усовершенствованный капельный метод single‑cell ATAC‑seq, использующий кастомные гидрогелевые бусины для баркодирования отдельных клеток. Они сравнивают HyDrop v2 с широко используемым коммерческим набором, создавая большие атласы на двух очень разных системах: моторной коре взрослого мыша и позднеэтапных эмбрионах дрозофилы. HyDrop v2 генерирует сопоставимое качество данных — выявляет те же основные типы клеток и очень похожие наборы доступных регионов ДНК — при этом обходится примерно в четырнадцать раз дешевле на образец мышиного мозга. Важно, что данные HyDrop v2 хорошо интегрируются с коммерческими данными, что позволяет исследователям комбинировать платформы при построении очень крупных атласов.
Обучение моделей глубокого обучения распознавать логику энхансеров
Чтобы проверить, достаточно ли дешёвых данных для продвинутого моделирования, команда обучает модели «от последовательности к функции» на атласах, полученных либо коммерчески, либо с помощью HyDrop v2. Эти модели учатся напрямую по последовательности ДНК предсказывать, насколько доступен каждый регион в каждом типе клеток, и могут выявлять короткие последовательностные мотивы, которые, вероятно, соответствуют сайтам связывания для конкретных регуляторных белков. В коре мыши модели, обученные на данных HyDrop v2, сопоставимы по общей точности с моделями на коммерческих данных и по способности восстанавливать известные «переключатели» энхансеров, ранее валидированные in vivo. В эмбрионе дрозофилы обе платформы поддерживают модели, которые могут анализировать 2000‑парные участки и выделять ключевые ~500‑парные сегменты, реально управляющие тканеспецифической активностью энхансеров, например регионы, контролирующие экспрессию генов нейробластов или мышц.
Больше клеток лучше глубины
Практический вопрос для любой лаборатории — стоит ли глубоко секвенировать каждую клетку или профильтровать больше клеток с меньшей глубиной. Путём систематического изменения числа клеток и числа фрагментов ДНК на клетку авторы показывают, что производительность моделей почти не страдает при снижении глубины секвенирования до умеренного уровня, если включено достаточное число клеток. Напротив, сокращение числа клеток явно ухудшает точность модели, особенно при оценке по множеству типов клеток одновременно. Поскольку HyDrop v2 намного дешевле на одну клетку, исследователи могут легко добавить десятки тысяч дополнительных клеток, восстанавливая или даже превосходя производительность моделей на коммерческих данных при небольшой доле затрат.
Наблюдая следы белков на ДНК
Исследование также проверяет, вводят ли разные лабораторные платформы тонкие смещения в том, как фермент ATAC‑seq разрезает ДНК, что могло бы вводить в заблуждение модели, пытающиеся определить, где на геноме находятся белки. С помощью отдельного нейросетевого инструмента, корректирующего предпочтения фермента, авторы показывают, что HyDrop v2 и коммерческие наборы дают практически идентичные шаблоны активности фермента как в мышиных, так и в дрозофиловых клетках. После коррекции оба набора данных раскрывают мелкомасштабные «следы», где регуляторные белки и нуклеосомы, по‑видимому, защищают ДНК от разрезов, и эти следы совпадают с последовательностными мотивами, выделяемыми моделями «от последовательности к функции». Такое согласие указывает, что как открытые, так и коммерческие платформы одинаково годятся для детальных исследований взаимодействия белков с ДНК.
Почему это важно для расшифровки генома
Для неспециалистов главный вывод в том, что теперь мы можем строить очень большие, доступные по стоимости карты того, как ДНК используется в отдельных клетках, и обучать на этих картах мощные модели глубокого обучения без необходимости полагаться исключительно на дорогое проприетарное оборудование. HyDrop v2 предоставляет данные, поддерживающие предсказание энхансеров, интерпретацию последовательностных мотивов и выявление участков связывания белков на уровне, сопоставимом с ведущими коммерческими методами, при условии профилирования достаточного числа клеток. Это открывает путь к созданию атласов регуляторных элементов по всему организму в здоровом и заболевшем состояниях, ускоряя исследования по чтению регуляторных инструкций генома и созданию новых, точно нацеленных генетических переключателей для науки и будущих терапий.
Цитирование: Dickmänken, H., Wojno, M., Mahieu, L. et al. Evaluating single-cell ATAC-seq atlasing technologies using sequence-to-function modeling. Nat Commun 17, 1951 (2026). https://doi.org/10.1038/s41467-026-68742-4
Ключевые слова: single-cell ATAC-seq, энхансеры, модели глубокого обучения, регуляция генов, открытая геномика