Clear Sky Science · ru
Альтернативная активация EGFR мутантой R252C из образца пациента способствует прогрессированию рака
Когда клеточные «антенны» выходят из-под контроля
Почему некоторые опухоли продолжают расти несмотря на курсы химиотерапии и передовую иммунотерапию? В этом исследовании прослеживается случай пациентa с опухолями в лёгком и мозге и восстанавливается история болезни до крошечной модификации в ключевой клеточной «антенне» — рецепторе EGFR. Выявив, как одна единственная мутация перенастраивает сигналы роста, авторы не только объясняют агрессивность болезни у пациента, но и показывают, что существующий препарат афатиниб способен сдерживать этот процесс.
Редкая мутация с большими последствиями
EGFR — это рецептор в мембране клетки, который помогает реагировать на сигналы роста. Во многих раках лёгкого и головного мозга встречаются мутации EGFR, но большинство известных изменений расположены внутри клетки, в участке, выполняющем роль ферментного переключателя. В этой работе команда обнаружила необычную замену снаружи EGFR, в области, которая обычно связывает факторы роста. У пациента с опухолями в лёгком и глиомой один аминокислотный остаток в позиции 252 был заменён из аргинина на цистеин — обозначено как EGFR R252C. Анализ баз данных по раку показал встречаемость этой мутации у небольшой доли пациентов с глиомой и почти никогда при опухолях лёгкого, что указывает на её редкость, но реальное существование. С помощью инструментов редактирования генома авторы воспроизвели эту точную мутацию в нескольких линиях клеток рака мозга и лёгкого человека, чтобы изучить её эффект.
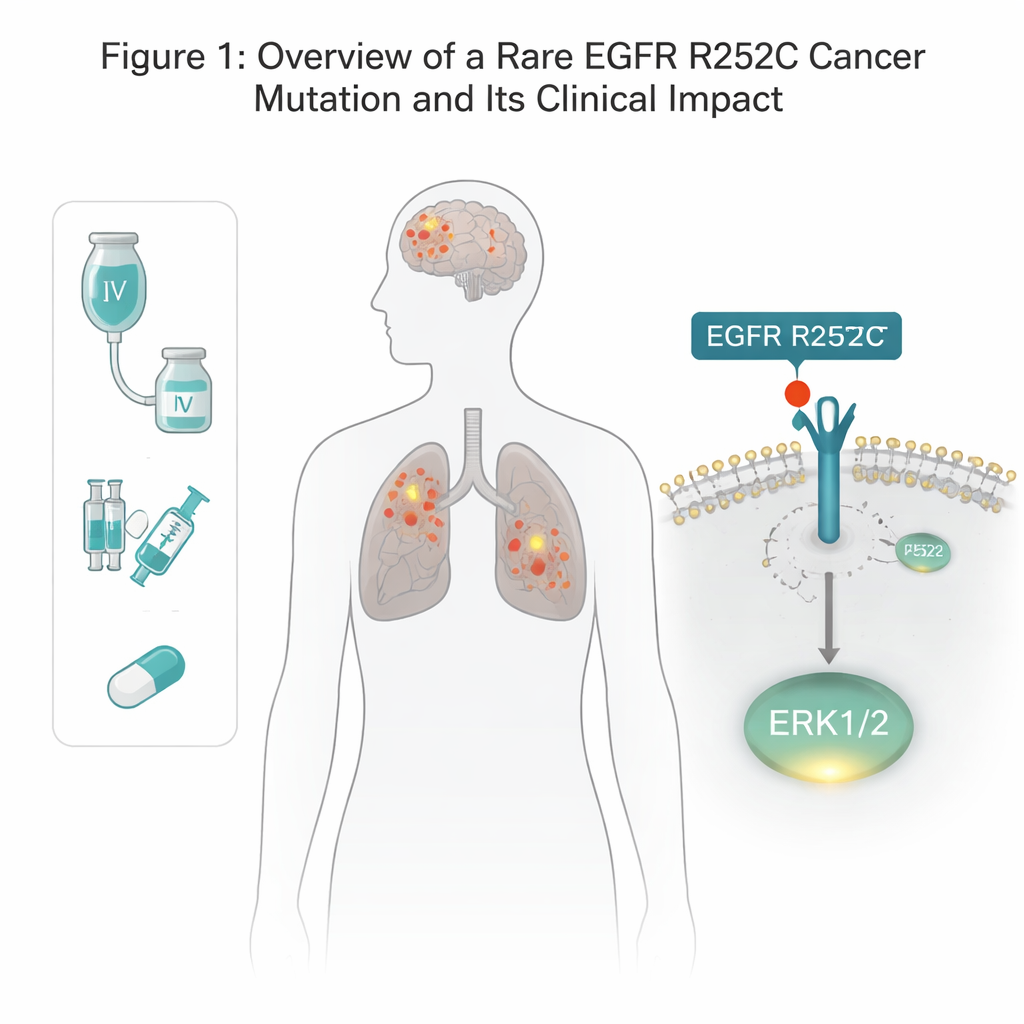
Новый обходной путь к сигналам роста
Обычно EGFR должен димеризоваться с другим копированием и затем фосфорилировать свою внутреннюю «хвостовую» часть, чтобы запустить сигнальные каскады роста. Удивительно, но версия R252C не демонстрировала типичной аутофосфорилирования. Вместо этого клетки с EGFR R252C значительно сильнее включали один конкретный регулятор роста — ERK1/2, в то время как другие классические пути EGFR, такие как AKT и STAT3, оставались по сути неизменными. Блокировка ERK1/2 специфическим ингибитором устраняла дополнительное преимущество в росте клеток R252C, доказав, что ERK1/2 является главным двигателем опухолевой активности этой мутации.
Фиксация рецептора в постоянно «включённой» форме
Чтобы понять, как внешнее изменение может вызывать такую селективную перегрузку, исследователи сочетали биохимические тесты с компьютерным моделированием. Замена на цистеин вводит новую цистеиновую группу на внешней части EGFR. Два таких мутантных рецептора могут образовать дисульфидный мост — своего рода молекулярную скрепку — между остатками C252, фиксируя их в стабильной паре. Структурное моделирование показало, что этот мост заставляет внешнюю часть рецептора принять «V-образное», со смещением выровненное положение, очень похожее на активную форму, связанную с лигандом, даже при отсутствии фактора роста. Это выравнивание передаётся через трансмембранный и подклеточный сегменты, скручивая внутренние ферментные домены в необычную конфигурацию: активные сайты обращены внутрь клетки, но находятся слишком далеко друг от друга для эффективного взаимного фосфорилирования. Вместо этого такая конформация создаёт сильную площадку для связывания ERK1/2, позволяя EGFR R252C напрямую фосфорилировать ERK1/2 и обходить обычную передачу через каскад RAS–RAF–MEK.
От моделей на мышах до одного пациента
Авторы показали, что клетки рака мозга и лёгкого с EGFR R252C росли быстрее в культуре и формировали большие, более агрессивные опухоли при трансплантации в мышей по сравнению с клетками с нормальным EGFR. Затем они испытали несколько поколений таблетированных ингибиторов EGFR. Только афатиниб, ингибитор второго поколения, последовательно выключал активацию ERK1/2 и резко снижал рост опухолевых клеток. В моделях на мышах с опухолями мозга и лёгкого, вызванными R252C, афатиниб замедлял рост опухолей и продлевал выживаемость. Критически важно, что когда исходный пациент, чьё заболевание прогрессировало несмотря на химиотерапию, препарат, нацеленный на кровеносные сосуды, и иммунотерапию, был переведен на афатиниб, КТ/МРТ обоих очагов показали заметное уменьшение опухолевой нагрузки, и пациент несколько лет жил без прогрессирования.
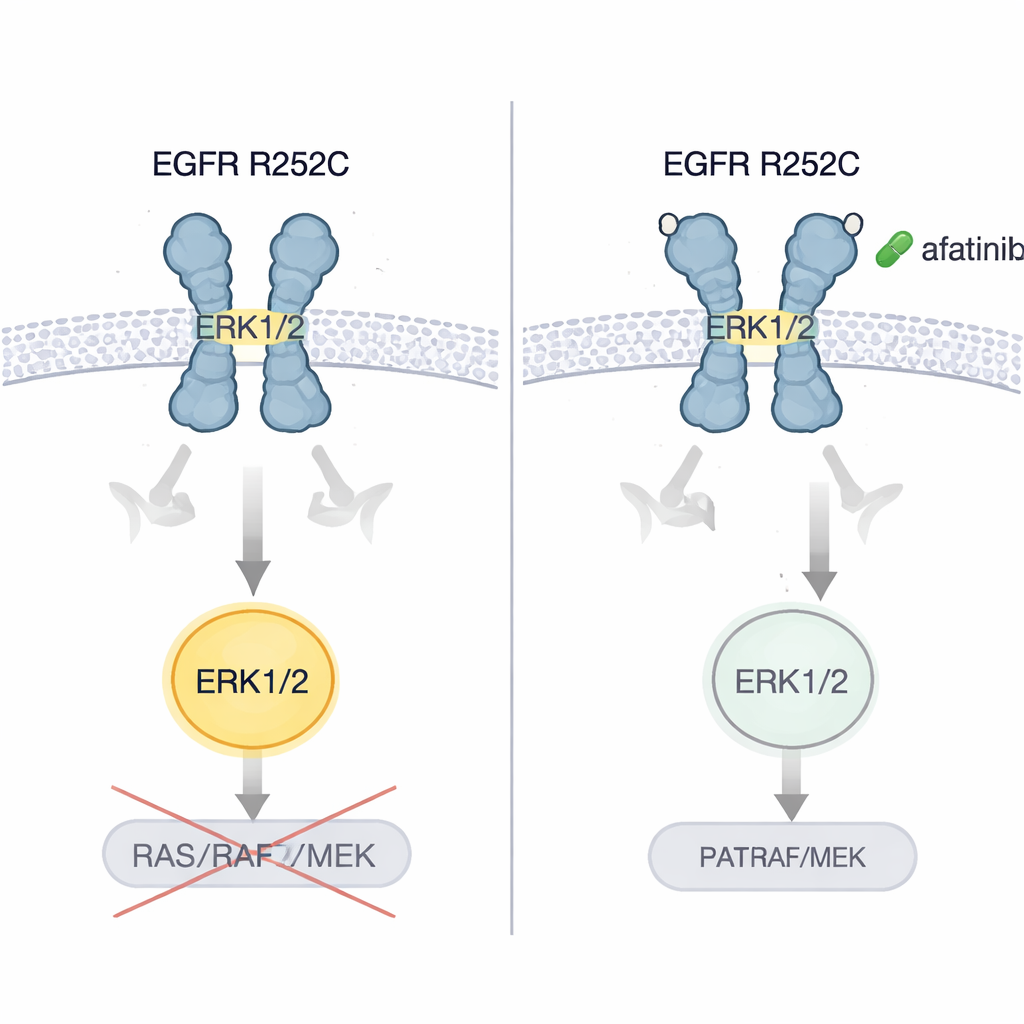
Что это значит для пациентов
Эта работа выявляет ранее нераспознанный механизм действия онкогенной мутации EGFR: скрепляя два рецептора вне клетки и закручивая их в активную позу, мутация напрямую включает ERK1/2 вместо прохождения по классической сигнальной цепочке. Для неспециалистов главный вывод таков: не все мутации в одном и том же гене ведут себя одинаково, и некоторые редкие изменения могут лучше отвечать на специфические уже существующие препараты. EGFR R252C, по-видимому, создаёт опухоли, особенно чувствительные к афатинибу. Хотя это заключение сейчас опирается на один подробный клинический случай и обширные лабораторные данные, оно указывает на необходимость более персонализированного тестирования мутаций внешнего домена EGFR и предполагает, что тщательно подобранные таргетные терапии могут дать новую надежду отдельным пациентам с трудно поддающимися лечению опухолями мозга и лёгкого.
Цитирование: Zhang, Y., Fei, Q., Li, Y. et al. An alternative EGFR activation by patient-derived R252C mutation promotes cancer progression. Nat Commun 17, 1902 (2026). https://doi.org/10.1038/s41467-026-68699-4
Ключевые слова: мутация EGFR, глиома, рак лёгкого, сигнализация ERK, афатиниб