Clear Sky Science · ru
Улучшение предсказаний по полиэнтным шкалам для недостаточно представленных групп с помощью transfer learning
Почему ваш генетический риск‑балл может вам не подойти
Генетические «риск‑баллы» всё чаще применяют для оценки вероятности развития распространённых заболеваний, таких как диабет, болезни сердца или гипертония. Но большинство таких шкал создано на данных ДНК людей европейского происхождения. В результате они часто дают плохие прогнозы для людей из других популяций, что вызывает вопросы справедливости и практической применимости в медицине. В этом исследовании ставится простой вопрос: можно ли повторно использовать знания, полученные на больших европейских наборах, чтобы построить более точные и справедливые генетические шкалы для недостаточно представленных групп — не обмениваясь при этом чьими‑то персональными данными?
От миллионов маркеров ДНК к единому баллу риска
Полиэнтный шкал — это как табель успеваемости, который суммирует небольшие эффекты множества генетических маркеров по всему геному. Каждому маркеру присваивается вес, отражающий силу его связи с признаком, на основе крупных генетических исследований. Когда эти исследования в основном включают европейцев, итоговая шкала работает лучше именно для европейцев. Различия в генетическом фоне — частотах вариантов ДНК и их совместном наследовании — означают, что те же веса часто не подходят афроамериканцам, латиноамериканцам и другим популяциям. Сбор одинаково больших наборов для каждой группы дорого и медленно, поэтому авторы обратились к стратегии машинного обучения, называемой transfer learning: вместо того чтобы начинать с нуля для каждой популяции, они уточняют уже существующую модель, обученную в другом месте.
Как заимствовать знания, не передавая сырые данные
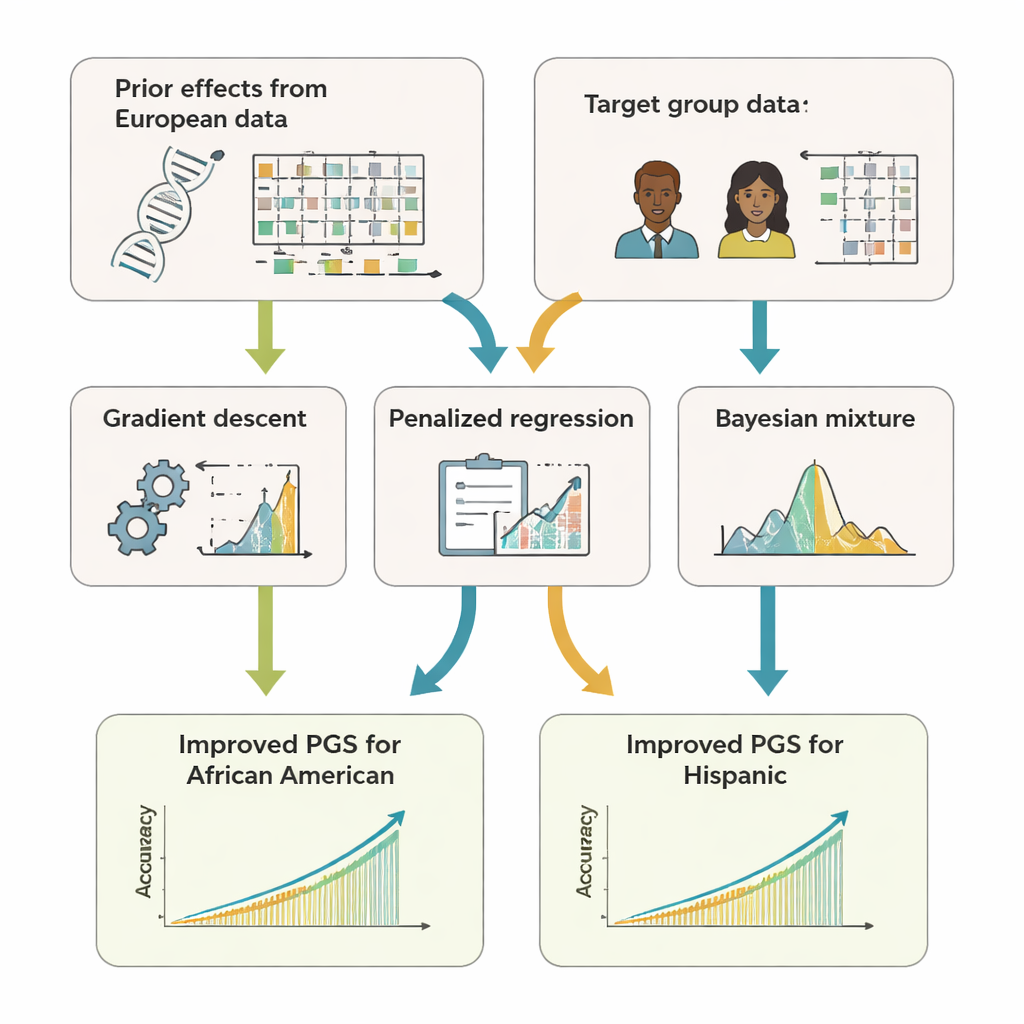
Команда разработала GPTL, пакет с открытым исходным кодом на R, реализующий три подхода transfer learning для генетических шкал. Все три метода стартуют от существующих оценок эффектов ДНК, полученных в большом наборе европейского происхождения, а затем мягко корректируют эти оценки на основе данных целевой группы, например афроамериканцев или латиноамериканцев. Один метод пошагово подправляет европейские веса с помощью градиентного спуска и останавливается рано, прежде чем полностью их заменить. Второй метод — штрафная регрессия — тянет новые оценки к исходным значениям, если только данные целевой группы не дают сильных доказательств обратного. Третий, байесовская смесь, позволяет каждому маркеру ДНК выбирать между несколькими источниками информации — например, разными группами по происхождению или даже вариантом «нет эффекта» — и смешивает их в зависимости от того, как хорошо они объясняют данные целевой группы.
Проверка методов на практике
Чтобы оценить эффективность подходов, авторы использовали как компьютерные симуляции, так и реальные данные сотен тысяч добровольцев из UK Biobank и американской программы All of Us. В качестве целевых групп они выбрали афроамериканцев и латиноамериканцев, а в качестве основного источника априорной информации — данные европейского происхождения. По результатам для 11 признаков, включая рост, индекс массы тела, липиды крови, кровяное давление и маркеры функции почек, transfer learning‑шкалы стабильно предсказывали лучше, чем шкалы, построенные исключительно на данных целевой группы или просто перенесённые из европейских наборов. Часто их точность соответствовала или немного превосходила более сложные «мульти‑происхождные» методы, требующие объединения сырых данных из нескольких популяций. Важный момент: методы GPTL нуждаются лишь в суммарной статистике — агрегированных числах об эффектах генов — поэтому учреждения могут сотрудничать, не раскрывая индивидуальные генетические записи.
Когда больше ДНК не всегда значит лучше
Исследователи также изучили, какие маркеры ДНК стоит включать. Вопреки распространённому мнению, что использование каждого доступного маркера всегда полезно, они обнаружили, что для афроамериканских и особенно латиноамериканских групп включение миллионов очень слабых сигналов может ухудшать качество, особенно при использовании сильно упрощённых представлений генетических корреляций. Сосредоточение на лучше обоснованных маркерах и использование более богатой информации о совместном наследовании вариантов часто давали более точные шкалы. Работа также показала, что добавление априорной информации из нескольких групп по происхождению и аккуратное моделирование различий между популяциями дополнительно улучшает предсказания.
Что это значит для более справедливых генетических прогнозов
Для неевропейских популяций стандартные генетические риск‑шкалы могут значительно проигрывать по точности, что потенциально усиливает неравенство в здравоохранении. Эта работа демонстрирует, что transfer learning — разумная адаптация европейских шкал с помощью умеренных наборов данных из недостаточно представленных групп — может сократить большую часть этого разрыва. На практике это означает, что клиники и исследователи могут строить более точные и справедливые генетические инструменты, не объединяя сырые данные между учреждениями или по происхождению, что снижает риски для конфиденциальности. Хотя ни один метод не будет оптимален для всех признаков и популяций, набор инструментов GPTL показывает, что технически достижимо сделать генетическое прогнозирование справедливее, если рассматривать прошлые модели не как конечный продукт, а как отправную точку для адаптации к разным группам.
Цитирование: Wu, H., Pérez-Rodríguez, P., Boehnke, M. et al. Improving polygenic score prediction for underrepresented groups through transfer learning. Nat Commun 17, 1973 (2026). https://doi.org/10.1038/s41467-026-68696-7
Ключевые слова: полиэнтные риск‑оценки, transfer learning, генетическое прогнозирование, неравенство в здравоохранении, популяционная генетика