Clear Sky Science · ru
TET1 как главный регулятор, контролирующий наблюдение за ферроптозом, зависимым и независимым от GPX4, при остром миелоидном лейкозе
Почему это исследование важно для лечения рака
Многие новые противораковые препараты направлены на то, чтобы заставить злонамеренные клетки запустить программу самоуничтожения — ферроптоз, форму гибели клеток, вызванную железом и повреждением жиров. Тем не менее некоторые опухоли упрямо сопротивляются этому подходу. В этом исследовании показано, как белок, модифицирующий ДНК, TET1 помогает лейкозным клеткам избегать ферроптоза через две отдельные биохимические системы защиты — и что блокирование этих защит может сделать даже резистентные опухоли уязвимыми.
Смертельная смесь железа и повреждённых липидов
Ферроптоз наступает, когда железо способствует неконтролируемому окислению липидов в мембранах клеток, что в конечном итоге приводит к их разрыву. При остром миелоидном лейкозе (ОМЛ), как и при многих других раках, клетки развивают мощные системы надзора, чтобы сдерживать этот процесс. Одним из ключевых защитников является фермент GPX4, который с помощью небольшого молекулярного носителя глутатиона нейтрализует вредные липидные пероксиды. Другие запасные системы генерируют антиоксидантные молекулы, способные улавливать опасные радикалы даже при нарушении GPX4. Понимание того, какие «главные переключатели» координируют эти защиты, важно для разработки терапий, которые надёжно индуцируют ферроптоз в раковых клетках, минимизируя вред здоровым тканям.
TET1 выступает центральным узлом управления
Исследователи сравнили десятки образцов раковых клеток, включая многие линии ОМЛ и клетки, полученные от пациентов, и заметили чёткую закономерность: клетки, устойчивые к ферроптозу, имели повышенные уровни TET1 — фермента, который изменяет химические метки ДНК и влияет на активность генов. Когда они снижали уровень TET1 с помощью генетических методов или ингибировали его активность малой молекулой, раковые клетки становились заметно более чувствительными к препаратам, индуцирующим ферроптоз. Это наблюдалось как в культурах клеток, так и в мышиных моделях ОМЛ. Напротив, повышение экспрессии TET1 защищало клетки от ферроптотической гибели и снижало накопление реактивных форм кислорода — агрессивных химических побочных продуктов, которые приводят к повреждению мембран.
Усиление основной антиоксидантной защиты
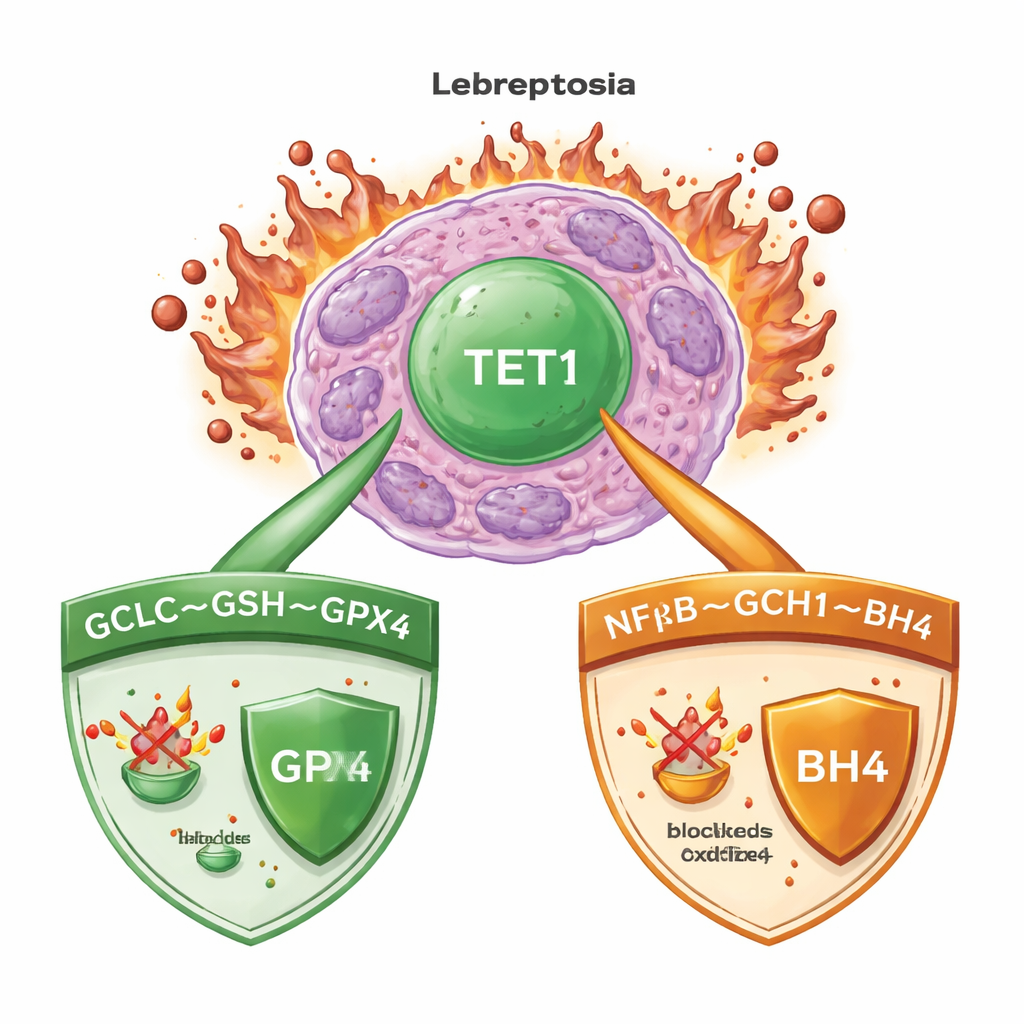
Углубив анализ, команда картировала участки генома, на которые действует TET1, и обнаружила, что он напрямую активирует ген GCLC. GCLC кодирует ключевой фермент, запускающий синтез глутатиона — «топлива» для GPX4. Увеличивая определённую метку ДНК (5-гидроксиметилцитозин) в промоторе GCLC, TET1 усиливает синтез глутатиона. При нормальных питательных условиях это увеличивает основной пул антиоксидантов клетки. При дефиците цистина тот же ферментный комплекс также синтезирует необычные γ-глутамил-пептиды, которые помогают убирать избыточный глутамат — ещё один способ ослабить ферроптоз. Как в клеточных культурах, так и у мышей потеря TET1 или фармакологическое подавление синтеза глутатиона резко снижали уровни глутатиона и этих защитных пептидов, делая лейкозные клетки значительно более уязвимыми к триггерам ферроптоза.
Второй путь побега, независимый от GPX4
Удивительно, но защитная роль TET1 не ограничивалась осью глутатион–GPX4. Даже при удалении GPX4 из лейкозных клеток повышенный уровень TET1 всё ещё мог предотвращать ферроптотическую гибель, что указывало на вторую линию защиты. Авторы проследили это до активации сигнального пути NFκB под воздействием TET1, в частности компонента NFKB2. Это, в свою очередь, усиливает экспрессию GCH1 — фермента, производящего антиоксидантную молекулу BH4. BH4 может защищать мембранные липиды от окисления без участия GPX4. При генетическом выключении или химическом блокировании GCH1 способность TET1 защищать клетки от ферроптоза частично терялась. В совокупности эти данные описывают путь TET1–NFKB2–GCH1, формирующий систему наблюдения за ферроптозом, независимую от GPX4.
Преобразование слабости в терапевтическую возможность
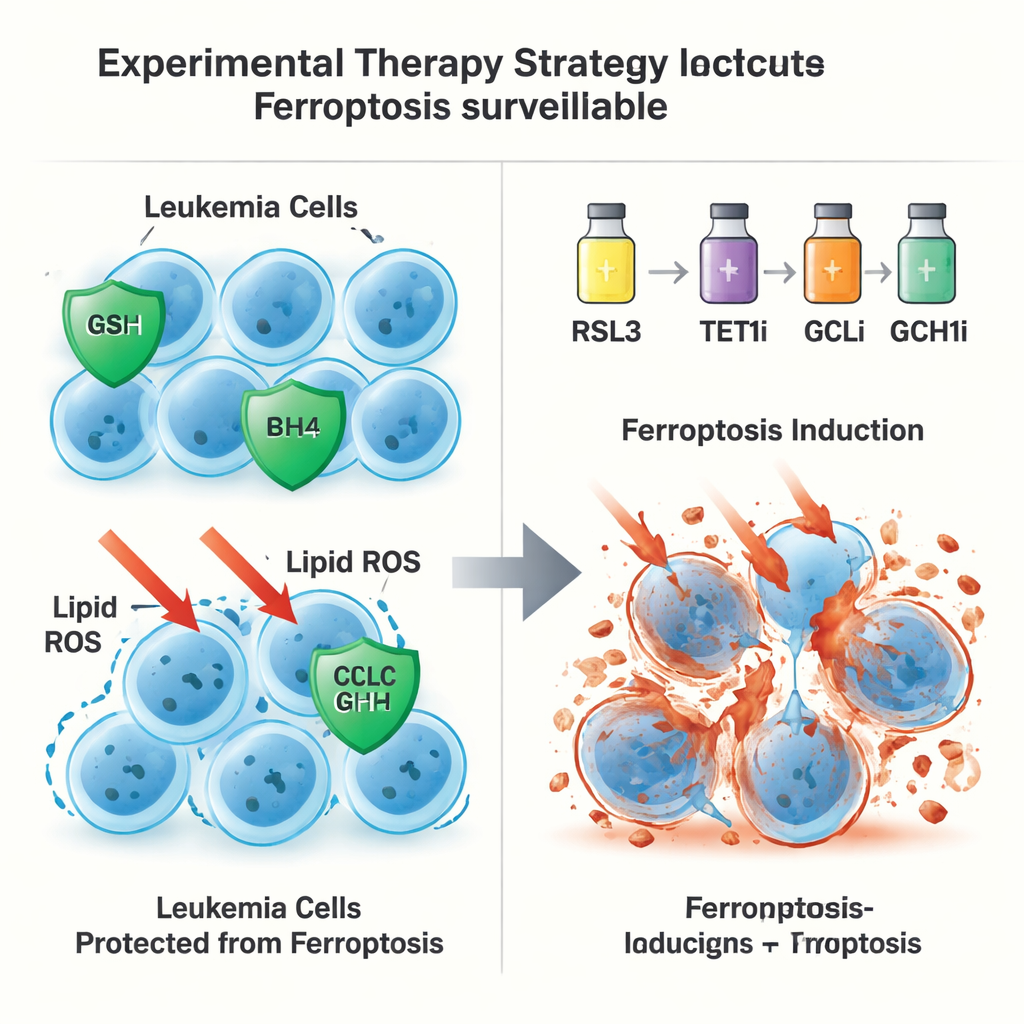
Имея карту двух путей, исследователи проверили, не даст ли преимущество одновременное стимулирование ферроптоза и ослабление контролируемых TET1 защит. В мышиных моделях ОМЛ и при трансплантациях лейкозных клеток, полученных от пациентов, низкие дозы препарата, индуцирующего ферроптоз, в сочетании с ингибиторами TET1, синтеза GSH (через GCLC) или GCH1 резко снижали нагрузку лейкоза, продлевали выживание и истощали популяции клеток, инициирующих лейкоз. Важно, что индуктор ферроптоза применялся в долях доз, описанных в предыдущих исследованиях, что снижает тревогу по поводу токсичности для нормальных кроветворных стволовых клеток.
Что это значит для будущих противораковых терапий
Для неспециалистов ключевой вывод таков: лейкозные клетки выживают, поддерживая две перекрывающиеся антиоксидантные «щиты», обе координируемые TET1 — один основан на глутатионе и GPX4, другой — на GCH1 и BH4. Работа показывает, что умеренное стимулирование ферроптоза при одновременном блокировании TET1 и его нисходящих партнёров может в перспективе позволить врачам преодолевать резистентность и селективно доводить раковые клетки до гибели, не подрывая здоровье нормальных тканей. Хотя эти стратегии ещё не готовы для клиники, исследование выделяет TET1 как мощный узел управления и перспективную мишень для комбинированных терапий при ОМЛ и, возможно, других трудноизлечимых раках.
Цитирование: Yang, L., Lu, J., Yun, W. et al. TET1 as a master regulator controlling GPX4-dependent and -independent ferroptosis surveillance in acute myeloid leukemia. Nat Commun 17, 1800 (2026). https://doi.org/10.1038/s41467-026-68509-x
Ключевые слова: ферроптоз, острый миелоидный лейкоз, TET1, глутатион, эпигенетика рака