Таргетные препараты революционизировали лечение некоторых форм рака легкого, нацеливаясь на дефектный сигнальный путь роста, называемый EGFR. Тем не менее у большинства пациентов эти лекарства перестают действовать в течение нескольких лет, поскольку опухоль развивается и приобретает устойчивость. В этом исследовании обнаружен неожиданный поворот: когда опухоли становятся резистентными к ингибиторам EGFR, у них возникает новая «ахиллесова пята», которую можно атаковать другим классом соединений. Понимание этой скрытой уязвимости может вдохновить будущие стратегии лечения, которые будут загонять эволюцию рака в тупик, а не постоянно ее догонять.
Обнаружена скрытая слабость
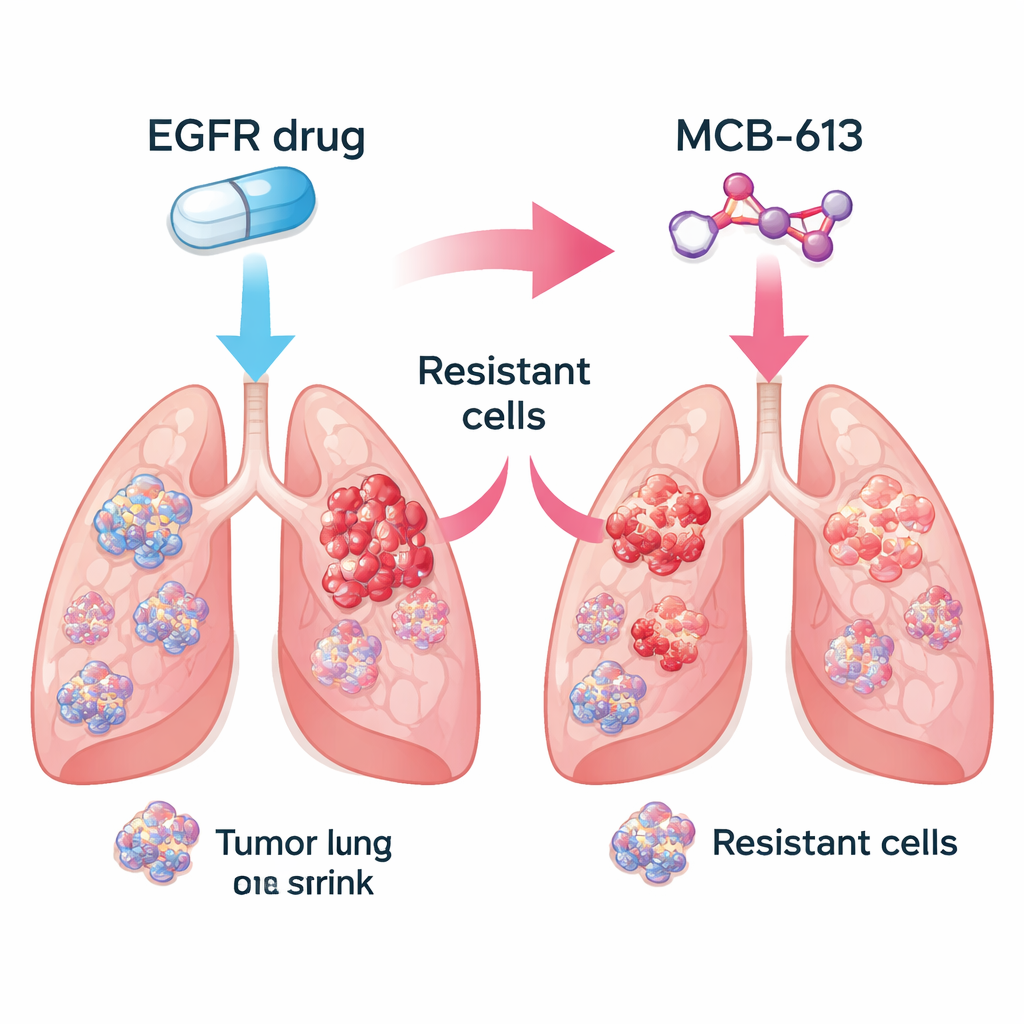
Исследователи сосредоточились на немелкоклеточном раке легкого, вызванном мутантным EGFR, распространенной форме заболевания. В лаборатории они сравнили чувствительные к лекарству раковые клетки с близкородственными клетками, которые эволюционно приобрели устойчивость к EGFR-ингибиторам, таким как гефитиниб и осимертиниб. Затем они протестировали библиотеку примерно из 2100 малых молекул, чтобы выяснить, какие из них убивают резистентные клетки эффективнее, чем исходные, чувствительные. Среди множества кандидатов одно соединение, названное MCB-613, последовательно выделялось. Клетки, игнорировавшие ингибиторы EGFR, оказались необычно уязвимы к MCB-613 как в культурах, так и в опухолях мышей.
Запирание смешанных популяций опухолей Figure 1.
Реальные опухоли представляют собой смеси клеток: некоторые остаются чувствительными к исходному препарату, в то время как другие приобретают устойчивость разными генетическими способами. Команда проверила, может ли сочетание ингибитора EGFR и MCB-613 уничтожить эту разношерстную популяцию. В контролируемом эксперименте они смешали в основном чувствительные клетки с небольшой долей нескольких типов резистентных клеток, имитируя опухоль пациента. Лечение этой смешанной популяции либо только ингибитором EGFR, либо только MCB-613 позволяло некоторым клеткам выжить и разрастаться. Но при одновременном применении обоих агентов вся популяция коллапсировала. Это свидетельствует о том, что сочетание стандартной таргетной терапии с тщательно подобранным препаратом «коллатеральной чувствительности» может загнать опухоль в эволюционный тупик.
Молекулярный мост, разрушающий стража
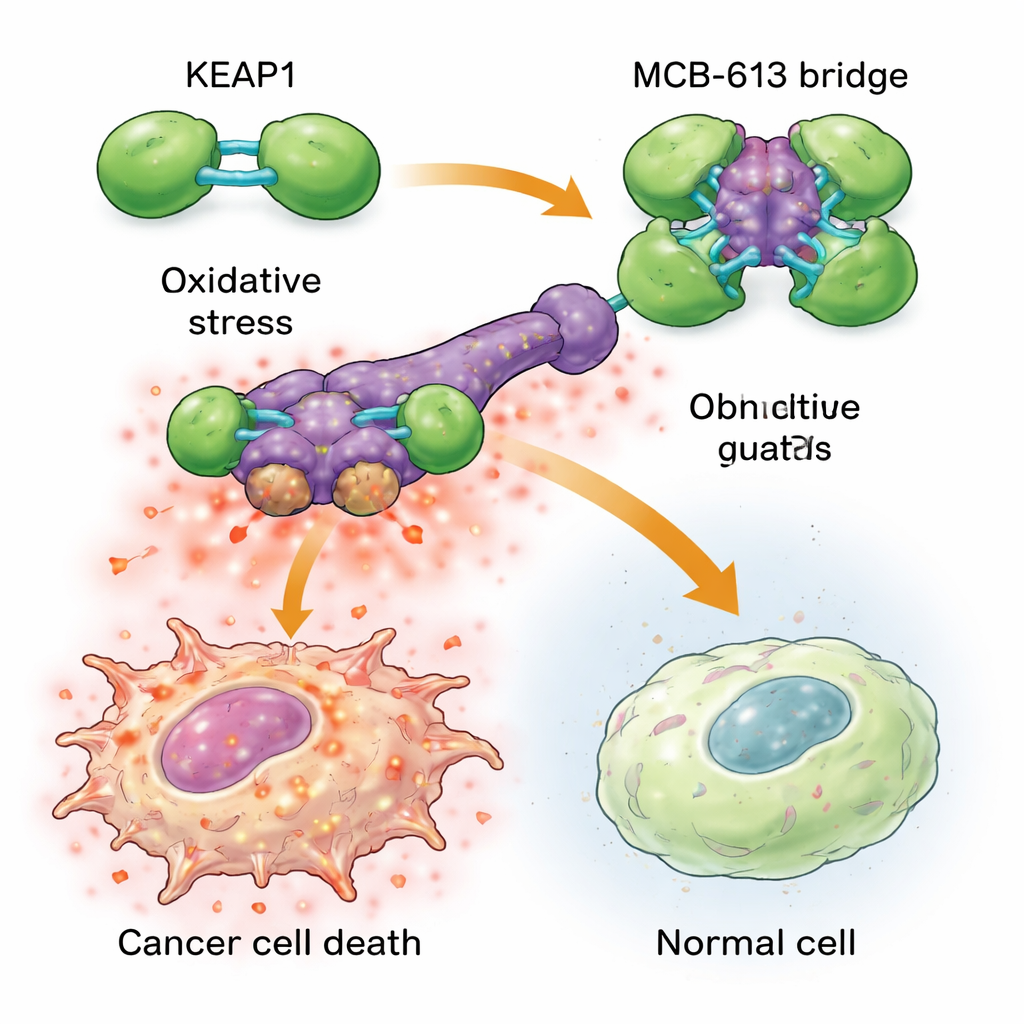
Чтобы понять, почему MCB-613 так сильно поражает резистентные клетки, ученые проанализировали белки, к которым оно связывается. С помощью химических зондов и таргетного CRISPR-скрининга они выделили белок KEAP1 как ключевой для действия MCB-613. Обычно KEAP1 действует как клеточный страж, распознающий стресс и регулирующий защитные ответы. Команда обнаружила, что MCB-613 присоединяется к KEAP1 необычным образом: он ведет себя как жесткий молекулярный мост, связывая молекулы KEAP1 в чрезмерные, аномальные кластеры. Этот процесс зависит не от привычных реактивных сульфгидрильных участков KEAP1, а от конкретного остатка лизина в области димеризации. Когда этот лизин мутировал, MCB-613 больше не мог кластеризовать KEAP1, и резистентные клетки утратили гиперчувствительность к соединению.
Превращение полезного стресса в смертельную перегрузку Figure 2.
Кластеризация KEAP1 запускает опасную цепную реакцию внутри лекарственно-устойчивых раковых клеток. Эти клетки уже существуют в условиях повышенного фонового стресса: у них повышены уровни реактивных форм кислорода (повреждающих побочных продуктов) и активность защитной сигнальной сети, известной как интегрированный ответ на стресс. При добавлении MCB-613 нарушение KEAP1 доводит это напряженное состояние до критического уровня: реактивные кислородные виды накапливаются еще сильнее, а ключевые регуляторы стресса ATF4 и CHOP запускают мощные программы гибели. Блокирование этих регуляторов стресса или химическое нейтрализование реактивных форм кислорода в значительной степени защищало клетки от MCB-613. Что интересно, классический партнер KEAP1 — NRF2, часто рассматриваемый как главный драйвер антиоксидантной защиты, не отвечал за гибель; напротив, удаление NRF2 делало клетки еще более чувствительными, подчеркивая, что MCB-613 эксплуатирует иной, неклассический путь.
Что это может значить для будущего лечения
Пока что сам MCB-613 является инструментальным соединением с химическими недостатками, делающими его непригодным в роли лекарства. Но он иллюстрирует мощную концепцию: по мере того как рак легкого развивается и становится устойчивым к ингибиторам EGFR, он может оказаться в фиксированном состоянии повышенного стресса, которое можно избирательно атаковать соединениями, заставляющими KEAP1 образовывать дисфункциональные сборки. В принципе, усовершенствованные версии таких «молекулярных мостов» можно было бы разработать более безопасными и точными, дав онкологам инструмент, чтобы загнать опухоль в «невозможный выбор» между чувствительностью к исходной таргетной терапии и чувствительностью к последующему агенту, индуцирующему стресс. Эта стратегия эволюционного запирания в перспективе может помочь отсрочить или преодолеть резистентность при раке легкого с мутацией EGFR и, возможно, при других трудноизлечимых опухолях.
Цитирование: Bassil, C.F., Dillon, K., Anderson, G.R. et al. EGFR inhibitor-resistant lung cancers exhibit collateral sensitivity to a covalent, cysteine-independent KEAP1 oligomerizing molecular bridge.
Nat Commun17, 1726 (2026). https://doi.org/10.1038/s41467-026-68424-1