Clear Sky Science · ru
Лучшие практики и инструменты в R и Python для статистической обработки и визуализации данных липидомики и метаболомики
Почему важно превращать лабораторные числа в понятные изображения
Современные приборы теперь способны измерять тысячи мельчайших молекул — липидов и других метаболитов — в одной капле крови или ткани. Эти измерения содержат подсказки о рисках заболеваний, ответах на лечение и о том, как наше тело реагирует на питание или возраст. Но необработанные данные не дают готового ответа: это огромная таблица чисел, которую нужно очистить, проанализировать и превратить в понятные визуализации. В этой статье объясняется, как исследователи могут использовать два популярных языка программирования, R и Python, чтобы делать это надежно, прозрачно и с графикой качества для публикаций.
От химических измерений к сложным таблицам данных
В липидомике и метаболомике масс-спектрометрия и хроматография генерируют большие наборы данных, где каждая строка — это образец, а каждый столбец — молекула. Эти таблицы редко ведут себя как аккуратные примеры из учебника. В них встречаются пропущенные значения, выбросы и скошенные распределения, где несколько молекул имеют чрезвычайно высокие уровни. Концентрации могут охватывать несколько порядков величины и влиять на результаты факторы, такие как возраст, пол, питание, лекарства, суточные ритмы, а также технические проблемы — дрейф прибора или батч-эффекты. Международные экспертные группы выпустили руководства по стандартизации сбора, обработки и отчетности по образцам, но даже при хорошей лабораторной практике тщательная статистическая обработка остается необходимой, чтобы выделить истинные биологические сигналы на фоне шума.
Очистка и подготовка чисел
Прежде чем сравнение между здоровыми и больными группами станет осмысленным, данные нужно подготовить. Обзор описывает, как появляются пропущенные значения — из-за случайных сбоев, ограничений прибора или помех в сигнале — и объясняет, когда их можно безопасно игнорировать, когда требуется повторное измерение и как их разумно оценивать (импутировать) с помощью методов, таких как k-ближайших соседей, случайные леса или простая подстановка низких значений. Далее авторы описывают стратегии нормализации, которые уменьшают нежелательную вариацию, например коррекцию батч-эффектов с использованием образцов контроля качества или корректировку на разницу в объеме проб. Затем обсуждаются преобразования, такие как логарифмы — которые устраняют длинные правые хвосты в распределениях — и методы масштабирования, которые приводят все молекулы к сопоставимому масштабу, чтобы очень изменчивые соединения не доминировали в последующем анализе. 
Статистические тесты и визуальные рассказы
После надлежащей подготовки данные поддаются ряду статистических инструментов. Для отдельных молекул исследователи могут вычислять относительные изменения (fold change) и использовать классические тесты, такие как t-тест, или их непараметрические аналоги (например, тест Манна — Уитни), чтобы выяснить, различаются ли уровни между группами. Для сравнения нескольких групп вводят методы, такие как ANOVA или тест Краскела — Уоллиса, сопровождаемые пост-хок процедурами для определения, какие именно группы отличаются. Мощь этих тестов раскрывается, когда их результаты визуализированы ясно. В статье выделяются коробчатые диаграммы (включая улучшенные версии для скошенных данных), виолончельные диаграммы и вулканические диаграммы, которые объединяют величину эффекта и статистическую значимость. Для липидов описываются более специализированные визуализации, например липидные сети, показывающие координированные изменения целых классов, и графики жирно-кислотных цепей, выявляющие закономерности в длине углеродной цепи и степени насыщения.
Видеть закономерности во множестве переменных одновременно
Поскольку каждый образец может иметь сотни или тысячи измеренных молекул, многомерные методы имеют решающее значение. Обзор объясняет, как анализ главных компонент (PCA) сжимает эту сложность в несколько новых осей, которые улавливают основные направления вариации, позволяя быстро проверять разделение групп, батч-эффекты или стабильность анализа. Более продвинутые нелинейные методы, включая t-SNE и UMAP, могут выявлять тонкие кластеры и структуры в высокоразмерном пространстве. В ситуациях, когда цель — классифицировать образцы, например отличать пациентов от контролей, авторы описывают контролируемые подходы на основе частичных наименьших квадратов и их ортогонального расширения (PLS-DA и OPLS-DA). Эти методы связывают молекулярные профили с метками образцов, поддерживают отбор признаков и часто суммируются с помощью графиков скорингов, загрузочных графиков и кривых приемник-оператор (ROC).
Практические наборы инструментов в R и Python
Чтобы помочь начинающим перейти от теории к практике, статья обзора рассматривает широкую экосистему программных пакетов. В R коллекции, такие как tidyverse и tidymodels, упрощают обработку данных и моделирование, тогда как ggplot2 и дополнительные пакеты вроде ggpubr, ggstatsplot и tidyplots облегчают создание иллюстраций публикационного качества. Специализированные библиотеки обрабатывают PCA, кластеризацию и модели на основе PLS, а пакеты Bioconductor поддерживают сложные тепловые карты и интерактивную графику. В Python pandas обеспечивает работу с таблицами, а matplotlib, seaborn и plotly покрывают визуализацию, а scikit-learn предлагает широкий набор многомерных методов. На протяжении всего текста авторы подчеркивают пошаговые примеры, доступные в сопутствующем GitBook, чтобы читатели могли воспроизводить рабочие процессы и адаптировать их к собственным данным. 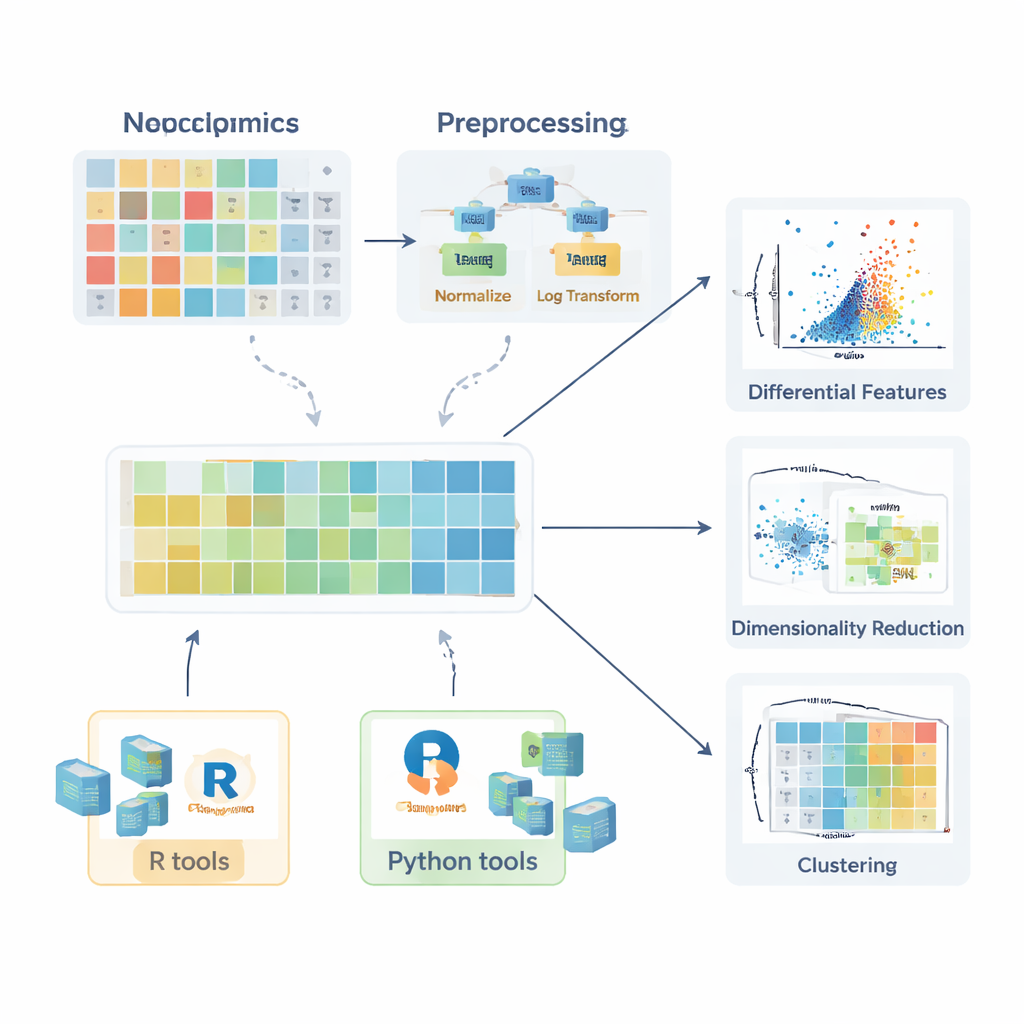
Превращение сложной химии в надежные выводы
В заключение авторы отмечают, что реальный потенциал липидомики и метаболомики заключается не только в мощных приборах, но и в том, насколько вдумчиво обрабатываются и визуализируются их результаты. Следуя хорошей статистической практике, используя открытые и хорошо документированные инструменты в R и Python и опираясь на общие примеры кода, исследователи могут строить надежные и воспроизводимые пайплайны. Это повышает шансы на то, что выявленные закономерности в мельчайших молекулах превратятся в достоверные биомаркеры, улучшат понимание механизмов заболеваний и приведут к более персонализированным подходам в медицине, что в конечном счете принесет пользу пациентам.
Цитирование: Idkowiak, J., Dehairs, J., Schwarzerová, J. et al. Best practices and tools in R and Python for statistical processing and visualization of lipidomics and metabolomics data. Nat Commun 16, 8714 (2025). https://doi.org/10.1038/s41467-025-63751-1
Ключевые слова: липидомика, метаболомика, визуализация данных, программирование на R, Python