Clear Sky Science · ru
Комбинирование пространственного транскриптома с морфологией ткани
Взгляд внутрь тканей двумя разными способами
Врачи и ученые всё чаще хотят знать не только какие гены активны в ткани, но и где именно они включены. В то же время в больницах повседневно получают детализированные изображения структуры ткани, которыми пользуются патологи. В этой статье объясняется, как новая область пытается связать эти два взгляда — подробные карты активности генов и обычные микроскопические снимки — и почему такое сочетание может привести к более ранней диагностике, лучшей градации рака и более глубокому пониманию того, как болезни развиваются и распространяются.
От разрозненных клеток к картам активности генов
В течение многих лет мощные «омиксные» методы требовали измельчения тканей до смеси одиночных клеток, что уничтожало информацию о том, откуда каждая клетка происходила. Пространственный транскриптом изменил это, измеряя активность генов при сохранении положения каждой клетки в ткани. В результате получается сетка точек, каждая со своим профилем экспрессии генов и точными координатами. Сама по себе такая пространственная информация о генах уже раскрыла новые закономерности клеточного разнообразия и архитектуры болезней. Но она неизменна после измерения, и повторный эксперимент обходится дорого. Напротив, изображения тканей, окрашенные стандартными красителями, такими как широко используемые гематоксилин и эозин (H&E), дешевы и доступны, и содержат визуальные подсказки о форме клеток, плотности и организации ткани.
Два способа объединить изображения и гены
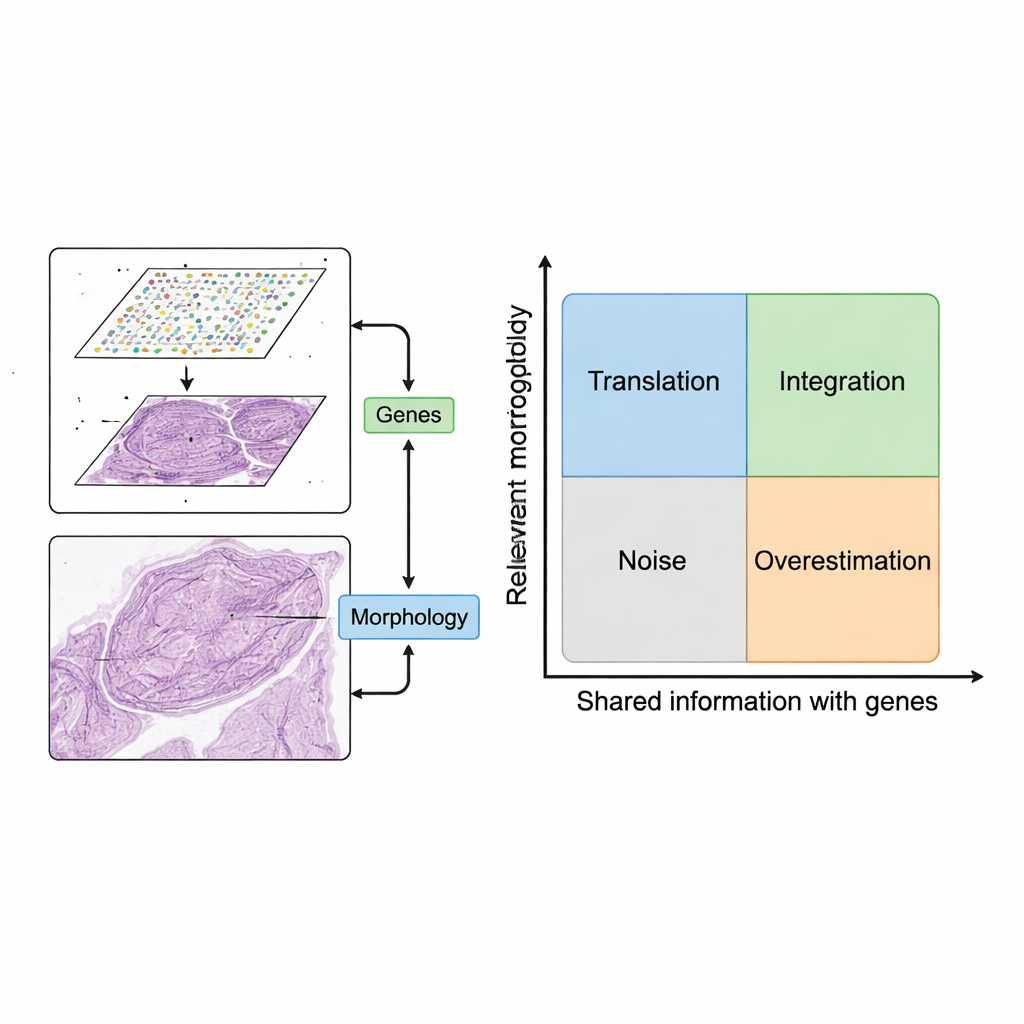
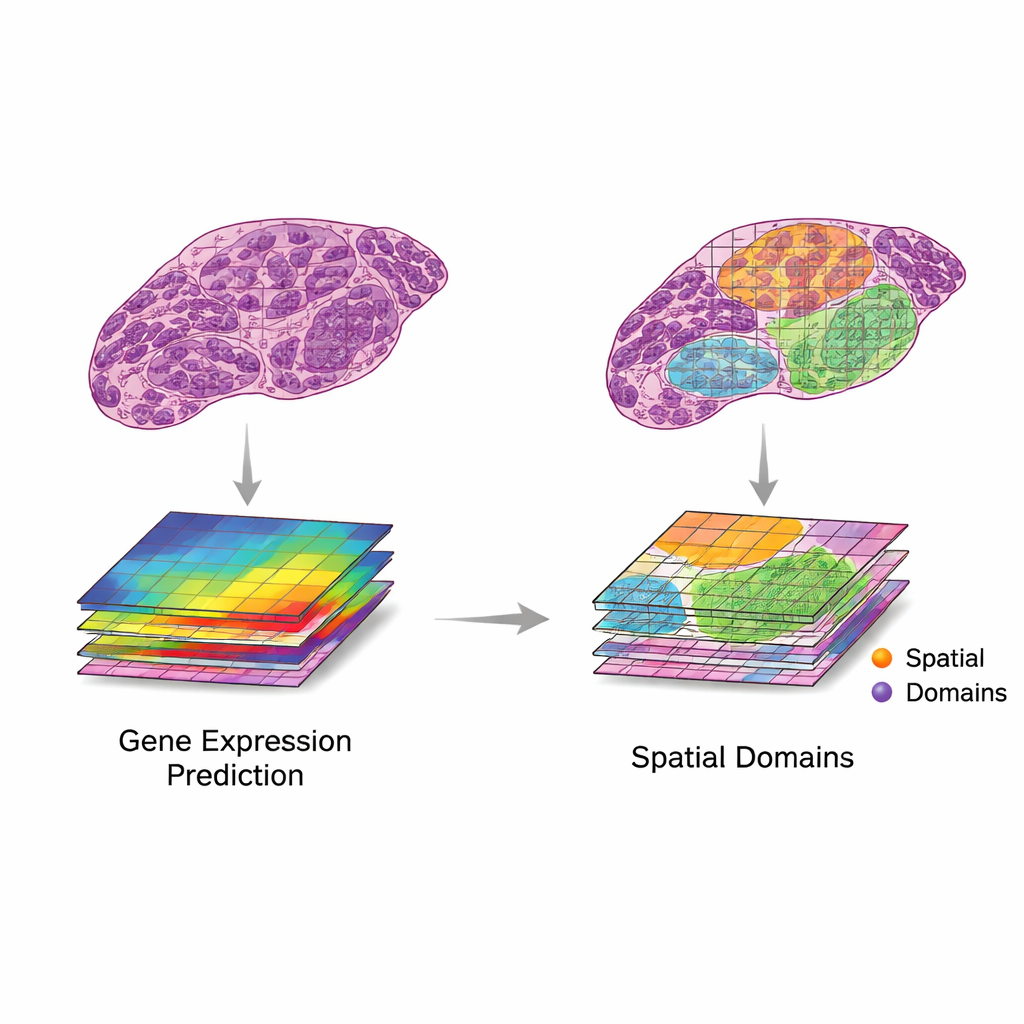
Обзор предлагает простую, но мощную схему использования этих двух источников данных вместе. Сначала фрагменты изображения сопоставляют с ближайшими точками экспрессии генов. Затем компьютерные модели извлекают признаки из изображений — шаблоны, отражающие форму, текстуру и организацию — и сравнивают их с паттернами в экспрессии генов. Авторы описывают два желательных сценария. В сценарии «трансляция» признаки изображения тесно соответствуют релевантной генетической активности, позволяя моделям предсказывать, какие гены включены, используя только изображение ткани. Это можно использовать, чтобы восстановить отсутствующие измерения генов, получить более высокое разрешение по сравнению с исходной сеткой или выводить активность генов по рутинным клиническим срезам без дополнительных лабораторных процедур. В сценарии «интеграция» признаки изображения улавливают полезную информацию, которой не хватает в генетических данных, например медленные структурные изменения или тонкую организацию ткани, помогая определить более чёткие регионы или «домены» внутри ткани.
Когда дополнительная информация помогает — и когда мешает
Не каждый признак изображения стоит использовать. Авторы вводят концептуальную карту с двумя осями: насколько признак изображения релевантен для биологического вопроса и насколько сильно он перекрывается с информацией о генах. Признаки, которые не относятся ни к релевантным, ни к генетическим аспектам, представляют собой шум, например артефакты окраски. Признаки, которые отслеживают генетические паттерны, но связаны с неважными генами (такими как базовые «домашние» гены), могут сделать модели внешне успешными, добавляя при этом мало клинической ценности. Организуя методы по четырём квадрантам — трансляция, интеграция, шум и переоценка — рамочная модель проясняет, когда объединение изображений и генов действительно добавляет понимание, а когда оно просто повторяет или затемняет уже известное.
Текущие инструменты, тесты и растущие боли
Быстро развивающаяся волна методов искусственного интеллекта теперь пытается выполнять трансляцию и интеграцию на реальных данных. Ранние системы опирались на сверточные нейронные сети, тогда как более новые используют трансформеры, графовые нейронные сети и многоуровневые модели, способные учитывать детали от мелких клеточных структур до контекста всего слайда. Эти методы применяли для предсказания активности генов по H&E-изображениям, генерации карт сверхвысокого разрешения и помощи в выделении областей ткани с отличающимся поведением. Для оценки качества исследователи опираются на статистические метрики, такие как корреляция между предсказанными и наблюдаемыми уровнями генов или согласованность между регионами, определёнными ИИ, и разметкой экспертных патологов. Однако наборы данных по‑прежнему малы и разнородны, и сравнить результаты между исследованиями трудно. Многие заявленные улучшения могут отражать переобучение или успех на генах и паттернах, имеющих мало значения в клинической практике.
К чему это может привести
Авторы приходят к выводу, что сочетание пространственных карт генов с изображениями ткани — перспективное, но всё ещё раннее направление. Современные модели часто достигают лишь умеренной точности и пока не готовы к рутинному медицинскому применению. Будущий прогресс, вероятно, придёт благодаря лучшим признакам изображений, в частности крупным «фоновым» моделям, обученным на миллионах патологических слайдов, и за счёт фокусировки на генах и паттернах, которые действительно влияют на уход за пациентом. Тщательно спроектированная интеграция однажды может выявлять ранние признаки болезни, обнаруживая несоответствия между тем, как ткань выглядит сейчас, и тем, что её гены предсказывают на будущее. Кратко говоря, эта работа прокладывает дорожную карту для превращения рутинных микроскопических изображений в богатые карты с информацией о генах, которые помогут врачам понимать и лечить болезни более точно.
Цитирование: Chelebian, E., Avenel, C. & Wählby, C. Combining spatial transcriptomics with tissue morphology. Nat Commun 16, 4452 (2025). https://doi.org/10.1038/s41467-025-58989-8
Ключевые слова: пространственный транскриптом, морфология ткани, цифровая патология, предсказание экспрессии генов, ИИ для изображений