Clear Sky Science · ru
Повышение активности KLF15 в кардиомиоцитах: новый подход к предотвращению патологической репрограммировки и фиброза с помощью нуклеазо-дефицитного dCas9VPR
Перепрограммирование больного сердца
Сердечная недостаточность затрагивает миллионы людей и часто развивается медленно после многих лет высокого артериального давления или заболеваний клапанов. При этих состояниях кардиомиоциты не только увеличиваются в размерах, но и активируют «фетальную» генетическую программу, а сердце заполняется рубцовой тканью. В этом исследовании изучается новый способ осторожно вернуть генетический контроль сердца в более здоровое состояние — без разрезания ДНК — путем умеренного увеличения уровня защитного регулятора KLF15 в кардиомиоцитах.
Когда клетки сердца теряют свою идентичность
В здоровом взрослом сердце кардиомиоциты — клетки сердечной мышцы — эффективно сжигают жиры для получения энергии и поддерживают стабильный профиль экспрессии генов. С помощью секвенирования РНК по отдельным клеткам у мышей, подвергнутых хронической перегрузке давления, исследователи проследили, как отдельные кардиомиоциты меняются по мере перехода сердца от нормальной функции к гипертрофии и затем к сердечной недостаточности. Они обнаружили, что транскрипционный фактор KLF15, который обычно поддерживает баланс метаболизма и роста, демонстрирует наиболее выраженное изменение активности в больных клетках. По мере увеличения стресса уровень KLF15 падал, и его способность сдерживать фетальные и стресс-индуцированные гены ослабевала. Похожие снижения KLF15 наблюдались и в человеческих сердцах пациентов с дилатационной и гипертрофической кардиомиопатией, что указывает на сохранность этого нарушения между видами.
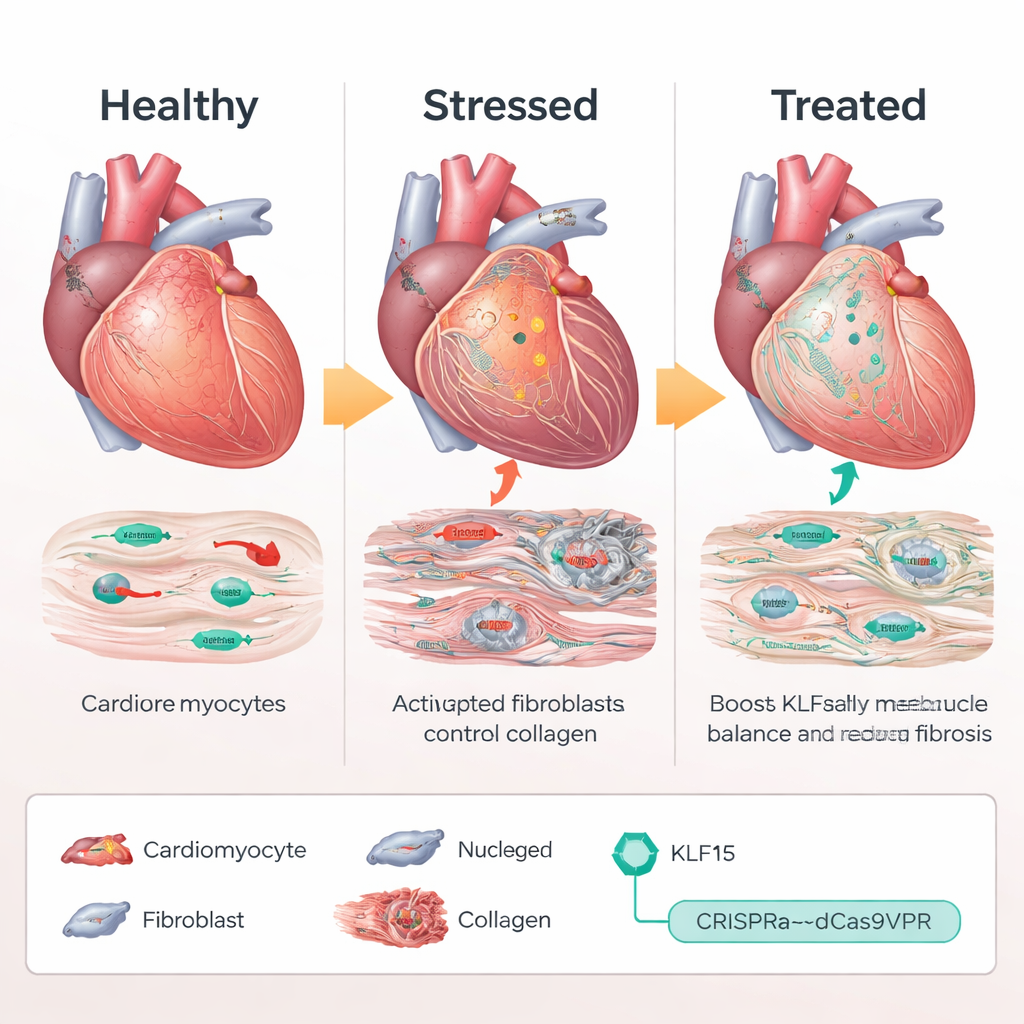
Использование CRISPR как регулятора громкости, а не как ножниц
Вместо того чтобы добавлять дополнительную копию гена KLF15 или разрезать ДНК, команда использовала систему «активации» на основе CRISPR, называемую dCas9VPR, которая связывается рядом с естественным геном Klf15 и усиливает его экспрессию. У мышей, генетически модифицированных так, чтобы этот CRISPR-активатор экспрессировался только в кардиомиоцитах, учёные доставили направляющие РНК с помощью адено-ассоциированного вируса (AAV9), нацеливая промотор Klf15. При хронической перегрузке давления у мышей, получивших направляющие, активирующие Klf15, уровни Klf15 оставались близкими к нормальным. Их кардиомиоциты оставались меньшими по размеру, снижение насосной функции было менее выражено, а выживаемость улучшилась по сравнению с контрольными животными. На молекулярном уровне гены, связанные со стрессом и фетальной программой, были подавлены, в то время как гены метаболизма и регуляции кальция восстанавливались, что указывает на значительное сбросывание вредной транскрипционной программы.
Торможение формирования рубца через клеточный кросс-ток
Сердечная недостаточность обусловлена не только больными кардиомиоцитами, но и фибробластами — поддерживающими клетками, которые вырабатывают коллаген и формируют жёсткую рубцовую ткань. Анализы на уровне отдельных клеток и тканевая визуализация показали, что восстановление Klf15 в кардиомиоцитах снижало активацию фибробластов и общую фиброзную перестройку, несмотря на то, что генная терапия напрямую не нацеливалась на фибробласты. Команда проследила этот эффект до секретируемого белка AZGP1. При усилении Klf15 в кардиомиоцитах производство и выделение AZGP1 увеличивались. Как в сердцах мышей, так и в тканях сердца, полученных из человеческих стволовых клеток, повышенный AZGP1 подавлял путь TGF-β / SMAD в фибробластах — ключевой драйвер образования рубцовой ткани — снижая уровни маркеров, таких как α-SMA и POSTN. Важно, что сверхэкспрессия AZGP1 только в кардиомиоцитах сама по себе не перепрограммировала мышечные клетки, что показывает: KLF15 прежде всего напрямую защищает кардиомиоциты и использует AZGP1 как посредника для сдерживания фибробластов.
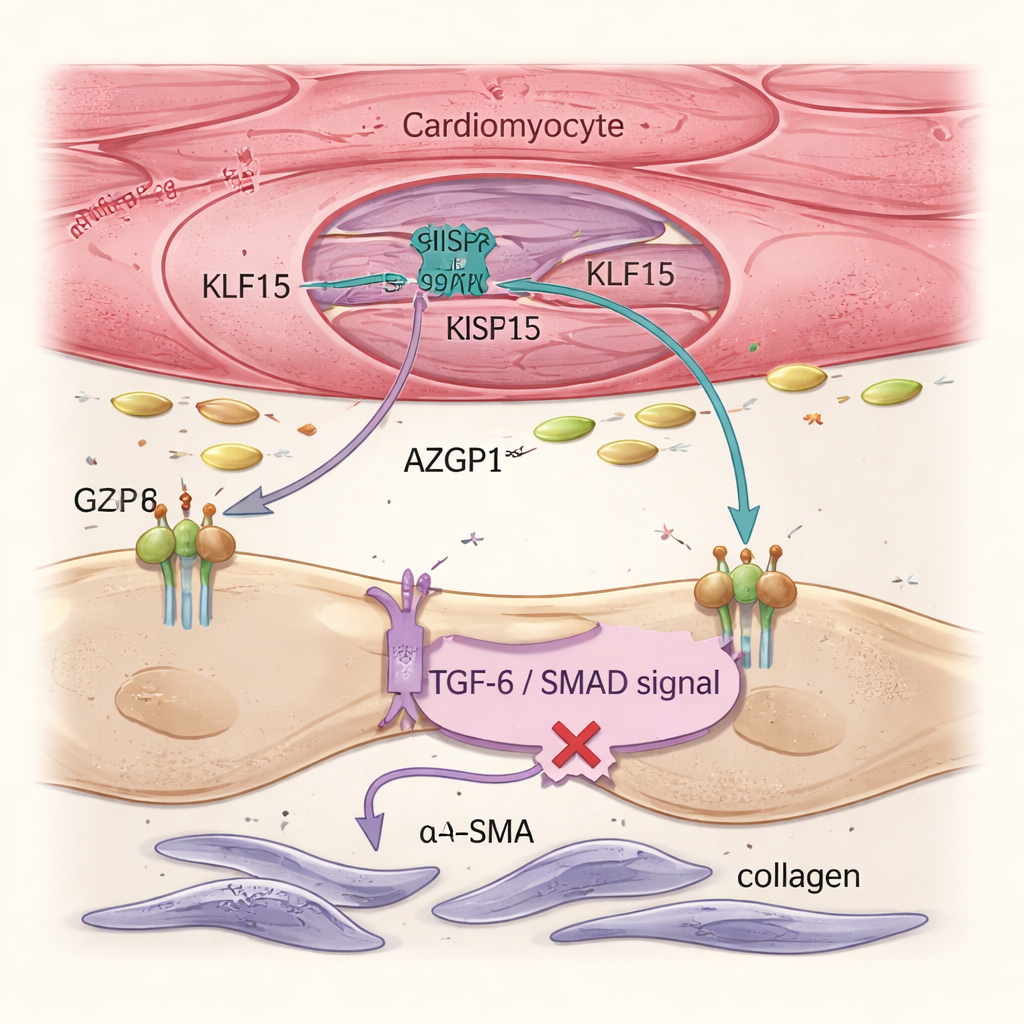
Модели человеческих тканей подтверждают защитную схему
Чтобы проверить, сохраняются ли эти механизмы в человеческих клетках, исследователи использовали кардиомиоциты, полученные из индуцированных плюрипотентных стволовых клеток, выращенные в трехмерных инженерных тканях сердца. При механической нагрузке, моделирующей высокое кровяное давление, эти ткани теряли KLF15, активировали стресс- и фетальные гены, уплотнялись, и их сокращения ослабевали — воспроизводя черты заболевания. Восстановление KLF15 с помощью CRISPRa предотвращало это ухудшение, сохраняло генерацию силы и возвращало экспрессию генов в сторону зрелого метаболизма и структуры. Подробные эксперименты показали, что TGF-β1, известный профибротический сигнал, снижает KLF15 в человеческих кардиомиоцитах через путь SMAD2/3, что помогает объяснить, как хронический стресс приводит к неблагоприятной ремоделировке. Наконец, команда создала компактную «мини»-систему CRISPRa на основе более мелкого варианта Cas9, которая помещается в один вектор AAV9 и управляется промотором, специфичным для кардиомиоцитов. В тонких срезах поражённой человеческой сердечной ткани этот вектор успешно повышал уровни KLF15 и в течение нескольких дней культивирования улучшал сократительную функцию.
План для более мягкой генетической терапии
Для неспециалиста суть такова: работа показывает, что аккуратное повышение одного защитного регулятора внутри кардиомиоцитов может одновременно стабилизировать их идентичность и посылать сигналы, ограничивающие образование рубцов. Используя активатор CRISPR, который не разрезает ДНК, подход тонко настраивает собственную генную программу сердца, а не вставляет искусственный ген. Исследование описывает путь TGF-β → KLF15 → AZGP1, связывающий механический стресс с вредной ремоделировкой, и демонстрирует на мышах, моделях человеческих клеток и срезах человеческой ткани, что восстановление KLF15 может разорвать эту цепную реакцию. Хотя это ещё доклиническая стадия, компактная, нацеленная на кардиомиоциты система CRISPRa, представленная здесь, предлагает потенциальную дорожную карту для лечения распространённых негенныхных форм сердечной недостаточности путём перепрограммирования активности генов, а не переписывания генома.
Цитирование: Schoger, E., Kim, R., Bleckwedel, F. et al. Enhancing KLF15 activity in cardiomyocytes: a novel approach to prevent pathological reprogramming and fibrosis via nuclease-deficient dCas9VPR. Sig Transduct Target Ther 11, 76 (2026). https://doi.org/10.1038/s41392-026-02593-9
Ключевые слова: сердечная недостаточность, KLF15, активация CRISPR, кардиальный фиброз, AZGP1