Clear Sky Science · pt
Acoplamento competitivo guiado por IA para triagem virtual e previsão de eficácia de compostos
Buscas mais Inteligentes por Novos Remédios
Encontrar novos medicamentos é um pouco como procurar uma agulha num palheiro formado por milhões de moléculas. Este estudo mostra como avanços recentes em inteligência artificial podem tornar essa busca mais rápida e barata, ajudando cientistas a prever quais moléculas têm maior probabilidade de se ligar a uma proteína associada à doença e realmente funcionar como remédio. Em vez de testar um químico de cada vez no laboratório, os autores usam modelos de IA para realizar competições virtuais entre moléculas e deixar que as vencedoras subam ao topo.
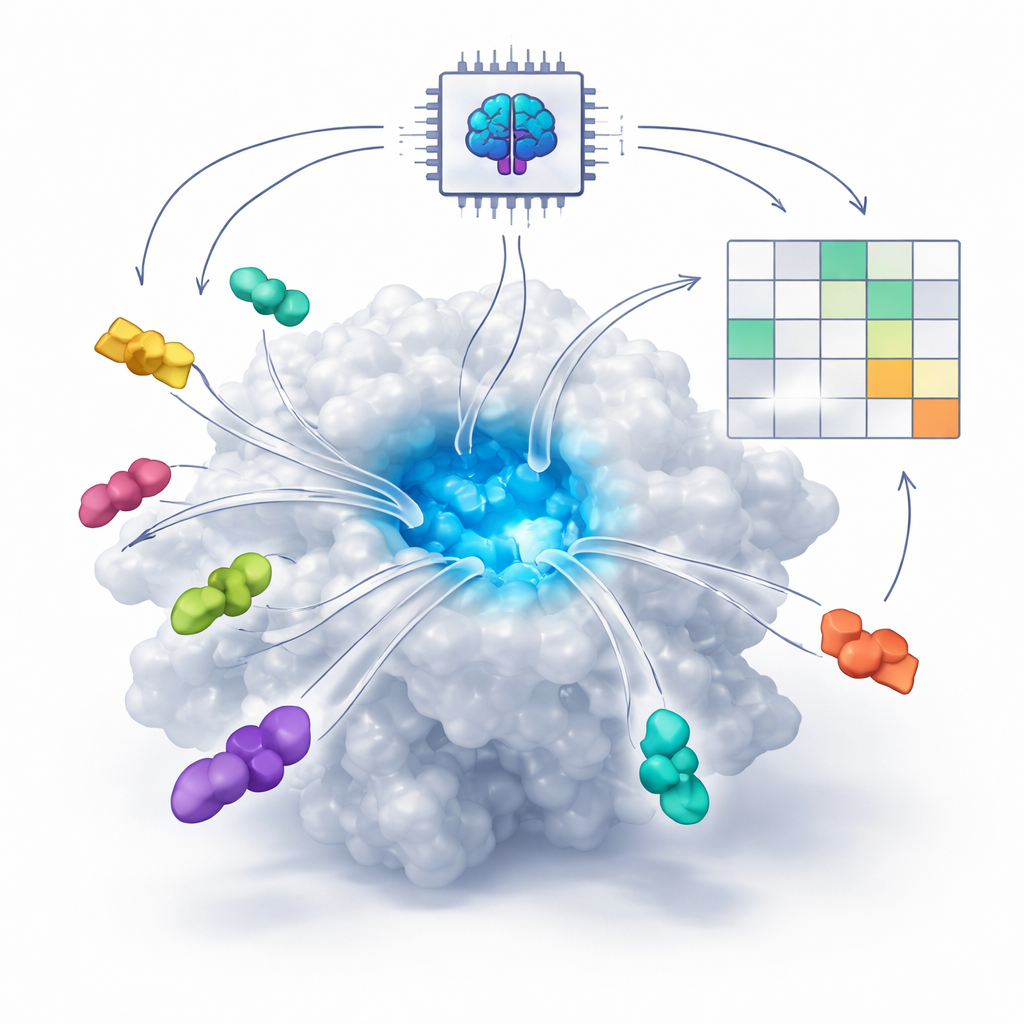
Como a IA Aprende a Ver Encaixes Tipo Fechadura-e-chave
Muitos fármacos modernos atuam se encaixando em bolsões minúsculos nas proteínas, tal como uma chave numa fechadura. Tradicionalmente, programas de computador tentavam prever esse encaixe usando equações físicas que estimam forças entre átomos. Nos últimos anos, porém, novos sistemas de IA chamados modelos de co-dobramento baseados em difusão — como AlphaFold3 e Boltz — aprenderam a partir de enormes números de estruturas conhecidas de proteína–molécula. Esses sistemas agora podem “imaginar” como uma proteína e um potencial fármaco podem se dobrar juntos em três dimensões, mesmo quando não existe uma estrutura experimental. A questão central que os autores abordam é se essas ferramentas de IA podem fazer mais do que apenas desenhar imagens plausíveis — se elas também conseguem distinguir bons fármacos dos ruins.
Ligantes Reais vs. Pretendentes
A equipe testou primeiro 16 proteínas bem estudadas além de uma enzima bacteriana mais complexa chamada DNA girase. Para cada proteína, pediram aos modelos de IA que posicionassem tanto inibidores ativos conhecidos quanto um conjunto de moléculas “fora do alvo” não relacionadas no mesmo sítio de ligação. Em vez de confiar numa única previsão, eles observaram com que consistência a IA colocava cada molécula em várias execuções. Inibidores verdadeiros tendiam a retornar ao mesmo ponto e orientação repetidas vezes, agrupando-se dentro de alguns trilionésimos de metro uns dos outros. Moléculas inativas vagavam mais amplamente e frequentemente ficavam mais distantes do bolso. Essa ideia simples — convergência de pose — revelou-se um sinal forte de que um composto realmente se ajusta ao seu alvo proteico.
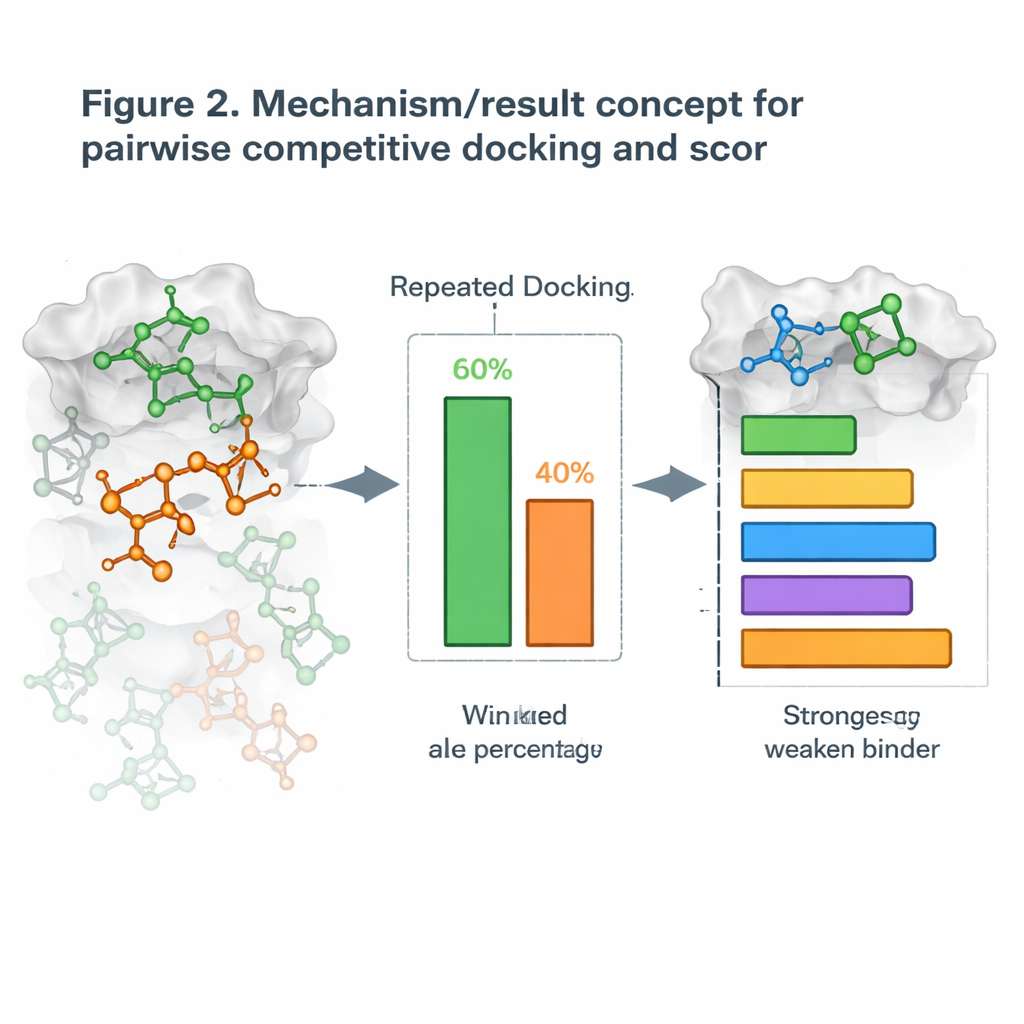
Transformando Docking em Competição Frente a Frente
Com base nisso, os autores introduziram uma nova estratégia que chamam de acoplamento competitivo pareado. Em vez de acoplar uma molécula por vez, eles acoplam dois candidatos simultaneamente com a proteína e os deixam “competir” pelo mesmo bolso. Após muitas execuções repetidas, a molécula que ocupa o sítio com mais frequência é declarada vencedora desse confronto. Ao executar todas as combinações possíveis, constroem uma tabela de vitórias–derrotas e calculam uma Pontuação de Acoplamento Competitivo para cada molécula, de forma semelhante ao ranking de jogadores em um torneio todos contra todos. Quando essas pontuações foram comparadas com medições do mundo real sobre o quão fortemente as moléculas bloqueiam seus alvos, as classificações frequentemente concordaram bem, com alguns sistemas proteicos apresentando quase acordo perfeito.
Da Triagem Virtual ao Desenho de Antibióticos Melhores
A DNA girase, uma enzima essencial para bactérias, serviu como um caso de teste detalhado. Essa proteína possui vários bolsões para drogas direcionados por diferentes classes de antibióticos, incluindo as amplamente usadas fluoroquinolonas. Os modelos de IA geralmente conseguiam posicionar cada classe de droga em seu bolso correto, e as pontuações de acoplamento competitivo acompanhavam aproximadamente suas potências medidas. Os autores então ampliaram para uma triagem virtual de mais de 3.000 medicamentos aprovados, perguntando quais moléculas competiam melhor pelo sítio das fluoroquinolonas. Sua estratégia em duas etapas — primeiro usando competição “todos-de-uma-vez” para escolher prováveis vencedores, depois filtrando pela firmeza com que se agrupavam no bolso — enriqueceu consideravelmente as verdadeiras fluoroquinolonas enquanto descartava candidatos mais fracos. Por fim, usaram um gerador de moléculas guiado por IA para propor novas estruturas semelhantes às fluoroquinolonas e aplicaram o acoplamento competitivo para encontrar um punhado com previsão de ligação ainda melhor e propriedades semelhantes às de fármacos aceitáveis.
Promessas, Limites e o que Isso Significa para Pacientes
O estudo mostra que modelos modernos de IA podem fazer mais do que desenhar estruturas proteína–droga plausíveis: quando executados em um quadro competitivo, eles podem ajudar a ranquear compostos de modo que muitas vezes espelha dados experimentais reais. Isso não substitui o trabalho de laboratório — o desempenho ainda depende fortemente da proteína em questão, alguns bolsões são mal previstos e modelos de IA podem falhar com moléculas muito grandes ou incomuns. Mas à medida que esses modelos e seus dados de treinamento melhorarem, abordagens como o acoplamento competitivo pareado podem tornar a descoberta inicial de fármacos muito mais eficiente. Para os pacientes, isso pode eventualmente se traduzir em desenvolvimento mais rápido de medicamentos direcionados, incluindo novos antibióticos que acompanhem bactérias resistentes.
Citação: Mirgaux, M., Barcelli, V., Chua, A.C.Y. et al. AI-guided competitive docking for virtual screening and compound efficacy prediction. npj Drug Discov. 3, 6 (2026). https://doi.org/10.1038/s44386-026-00039-4
Palavras-chave: descoberta de fármacos por IA, triagem virtual, acoplamento molecular, ligação proteína-ligante, desenho de antibióticos