Clear Sky Science · pt
Uma estrutura de ponta a ponta para reatividade em catálise heterogênea
Por que acelerar o desenho de catalisadores importa
A sociedade moderna depende de catalisadores para produzir combustíveis, plásticos, fertilizantes e inúmeros produtos do dia a dia. Ainda assim, encontrar catalisadores melhores costuma ser como procurar uma agulha num palheiro, porque cada material pode promover milhares de reações microscópicas possíveis simultaneamente. Este artigo apresenta o CARE, uma nova estrutura computacional que usa regras inteligentes e aprendizado de máquina para mapear e simular essas teias reacionais complexas muito mais rápido e de forma mais completa do que antes. Ao fazê-lo, promete orientar tecnologias de energia mais limpa e processos químicos mais eficientes, reduzindo drasticamente os custos computacionais.
Desembaraçando vias reacionais congestionadas
Na superfície de um catalisador sólido, moléculas que chegam não seguem simplesmente uma única rota direta do reagente ao produto. Em vez disso, elas atravessam um labirinto de intermediários de curta duração e vias concorrentes. Métodos computacionais tradicionais dependem da intuição humana para escolher um conjunto limitado de passos possíveis e então usam cálculos quânticos para avaliar suas energias. Isso funciona para redes pequenas, mas rapidamente falha à medida que os sistemas se tornam mais complexos, deixando de lado rotas raras que podem governar a atividade de longo prazo, a desativação ou a seletividade. O CARE enfrenta esse desafio construindo automaticamente redes reacionais muito grandes a partir de regras simples de montagem, garantindo que todos os eventos plausíveis de quebra e formação de ligações entre carbono, hidrogênio e oxigênio sejam incluídos, até aqueles que químicos normalmente poderiam descartar.
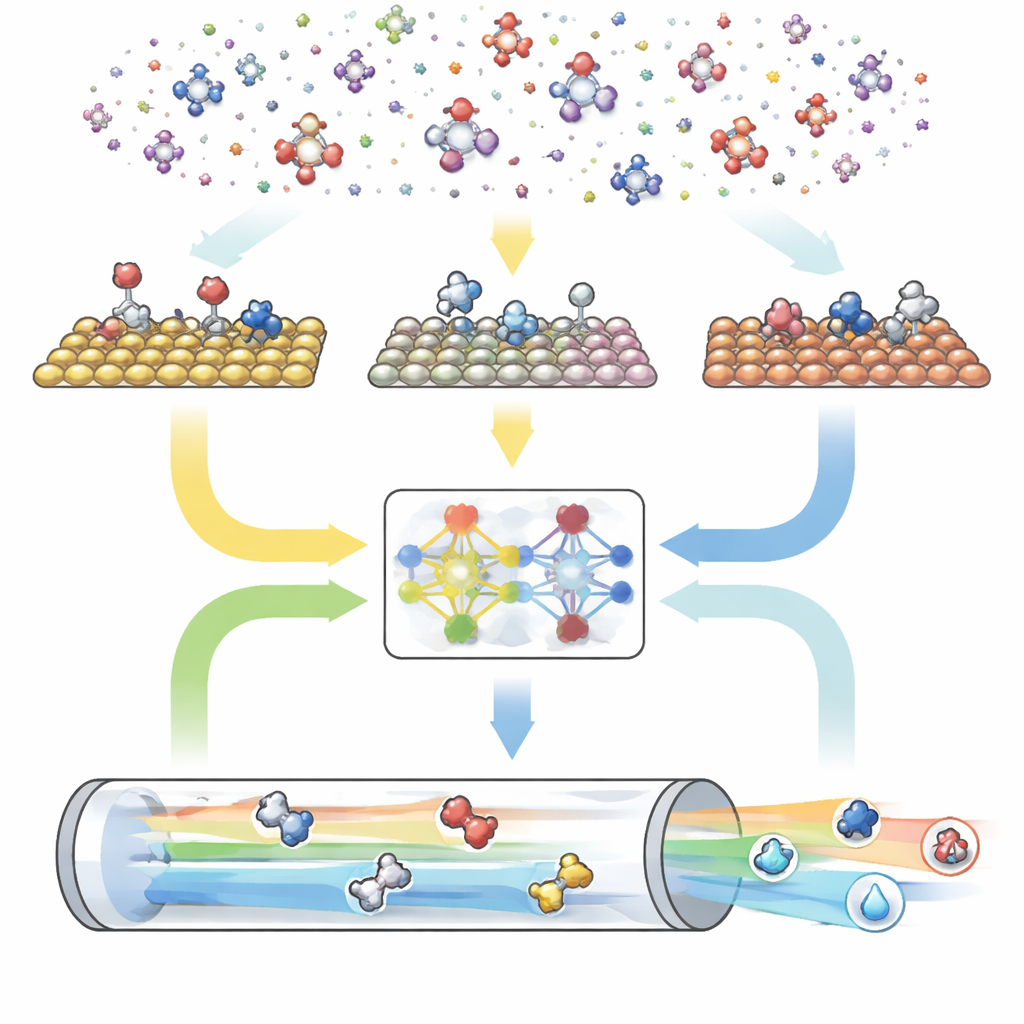
Um motor digital em três partes para reações
O CARE é organizado como um fluxo ponta a ponta com três módulos principais. Primeiro, um gerador baseado em regras define o “espaço químico” ao escolher o número máximo de átomos de carbono e oxigênio e então aplicar templates simples para criar todas as moléculas correspondentes e suas formas adsorvidas na superfície. Segundo, um módulo de avaliação de energias aciona modelos modernos de aprendizado de máquina — em especial uma rede neural gráfica chamada GAME-Net-UQ — para estimar as energias de intermediários e estados de transição em várias superfícies metálicas. Esse modelo trata cada estrutura como uma rede de átomos e ligações, devolve tanto uma energia quanto uma incerteza, e tem precisão dentro de alguns décimos de elétron-volt ao mesmo tempo em que é leve e rápido. Terceiro, um solucionador microcinético usa essas energias para calcular como todas as reações progridem em conjunto sob condições realistas de temperatura, pressão, potencial e pH, prevendo taxas reacionais globais, coberturas de superfície e seletividade de produtos.
Testes no mundo real: moléculas de combustível e química climática
Para mostrar que o CARE não é apenas um exercício teórico, os autores o aplicam a três problemas industriais relevantes de dificuldade crescente. Para a decomposição do metanol — uma reação importante para armazenamento de hidrogênio — eles geram uma rede modesta e a avaliam em muitos catalisadores metálicos e faces cristalinas. O CARE reproduz a conhecida tendência em “volcano” da atividade e identifica corretamente o rutênio como um dos melhores desempenhos, em concordância com experimentos, mas com uma fração ínfima do tempo de computação necessário para cálculos quânticos completos. Em seguida, avançam para a conversão eletroquímica de dióxido de carbono em cobre, focando em como produtos de três carbonos, como 1-propanol e propileno, surgem. Ao incluir passos especiais que tratam prótons, elétrons e condições de solução, o CARE captura como pH e potencial aplicado deslocam vias e prevê corretamente que 1-propanol é favorecido em relação ao propileno, ecoando estudos detalhados anteriores.
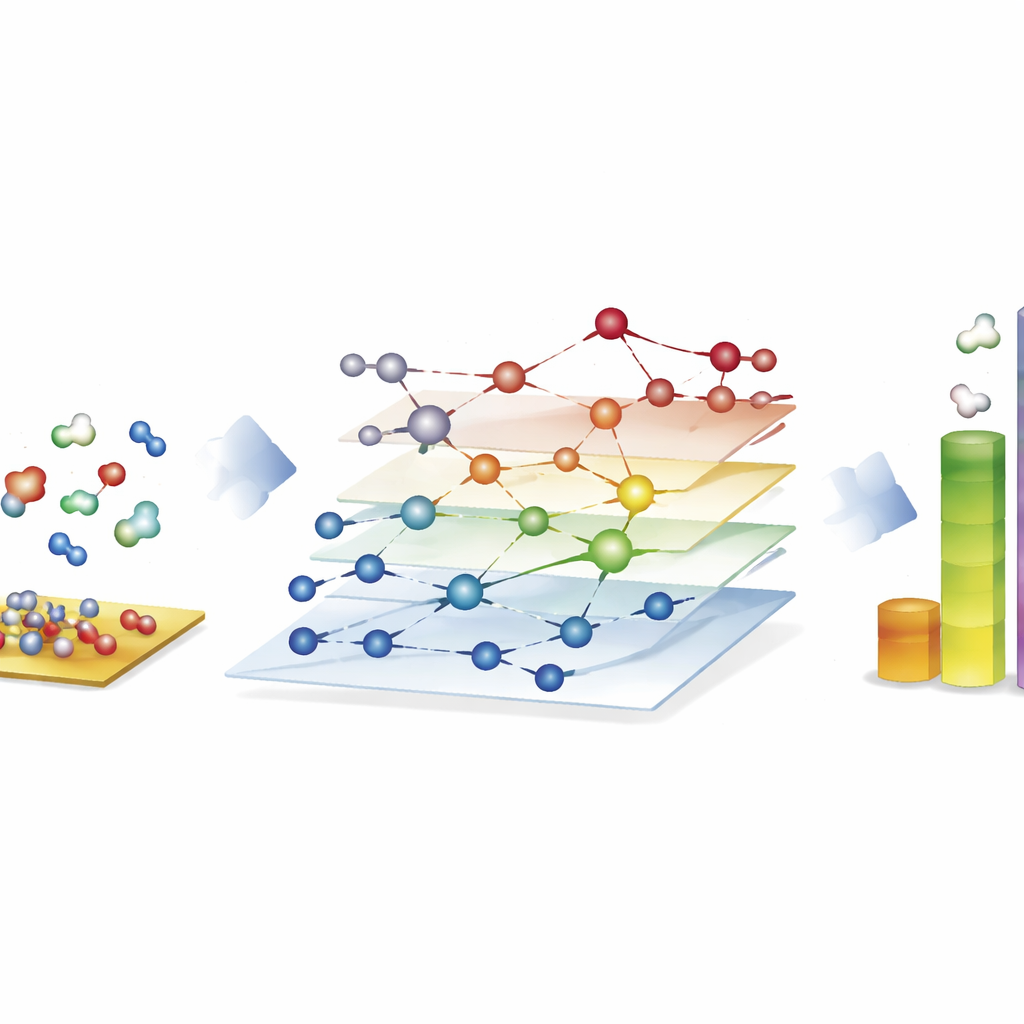
Explorando enormes redes reacionais para combustíveis sintéticos
A demonstração mais impressionante vem do processo de Fischer–Tropsch, que converte misturas de monóxido de carbono e hidrogênio em hidrocarbonetos de cadeia longa para combustíveis e produtos químicos. Aqui, os autores constroem redes com quase 40.000 espécies superficiais e cerca de 370.000 reações elementares — muito além do que estudos tradicionais baseados em mecânica quântica podem explorar totalmente. Usando o CARE, eles avaliam todos os intermediários e barreiras reacionais-chave em superfícies de cobalto, ferro, níquel e rutênio em apenas algumas horas em hardware padrão, uma aceleração de cerca de um milhão de vezes em comparação com cálculos quânticos diretos. Simulações microcinéticas nessas redes reproduzem tendências conhecidas: cobalto e ferro formam preferencialmente cadeias hidrocarbônicas mais longas, ferro produz mais dióxido de carbono por reações laterais, e níquel tende a maior hidrogenação. Embora alguns detalhes, como rendimentos de metano, permaneçam imperfeitos, a estrutura revela quais etapas de formação de ligações dominam o crescimento de cadeia e destaca onde os modelos ainda exigem refinamento.
O que isso significa para catalisadores futuros
Para não especialistas, a mensagem principal é que o CARE fornece um caminho prático para explorar espaços reacionais enormes em superfícies catalíticas que antes estavam fora de alcance. Ao automatizar a geração de redes, conectar modelos rápidos de aprendizado de máquina como “surrogatos” para energias quânticas e resolver a cinética resultante de forma eficiente, ele pode ranquear catalisadores candidatos, identificar condições operacionais promissoras e descobrir vias inesperadas com muito menos viés humano e custo computacional. Embora os autores reconheçam desafios remanescentes — como melhor tratamento de superfícies congestionadas, efeitos de solvente e redes ainda maiores — o trabalho aponta para um futuro em que computadores possam rastrear rapidamente reações complexas, desde redução de dióxido de carbono até reciclagem de plástico e upgrading de biomassa, orientando experimentos para as ideias mais promissoras em vez de deixar a descoberta ao acaso.
Citação: Morandi, S., Loveday, O., Renningholtz, T. et al. An end-to-end framework for reactivity in heterogeneous catalysis. Nat Chem Eng 3, 169–180 (2026). https://doi.org/10.1038/s44286-026-00361-8
Palavras-chave: catálise heterogênea, redes de reação, aprendizado de máquina, modelagem microcinética, síntese de Fischer–Tropsch