Clear Sky Science · pt
Difusão condicional com alinhamento modal sensível à localidade para gerar conjuntos conformacionais protéicos diversos
Por que o movimento das proteínas importa
As proteínas em nossas células não são esculturas rígidas; comportam-se mais como pequenas máquinas flexíveis que mudam constantemente de forma. Essas mudanças conformacionais podem controlar como enzimas catalisam reações, como receptores respondem a fármacos e como sinais se propagam pelas células. Ainda assim, a maioria das imagens familiares de proteínas mostra apenas uma “foto” da estrutura, sem refletir o rico conjunto de formas que realmente existem. Este artigo apresenta o Mac-Diff, um método de inteligência artificial que pode gerar rapidamente muitas formas realistas para uma dada proteína, ajudando os cientistas a ver não apenas como uma proteína é, mas como ela respira e se movimenta.
De instantâneos únicos a conjuntos em movimento
Durante décadas, pesquisadores dependeram de experimentos minuciosos ou de longas simulações de dinâmica molecular para explorar o movimento das proteínas, ambos lentos e caros. Ferramentas revolucionárias como o AlphaFold2 agora prevêem a estrutura 3D mais provável de uma proteína diretamente a partir da sequência de aminoácidos, mas normalmente retornam apenas uma ou poucas conformações preferidas. Muitas proteínas, especialmente as envolvidas em sinalização e regulação alostérica, naturalmente ocupam vários estados vagamente definidos. Os autores argumentam que, para entender como essas proteínas realmente funcionam — e para projetar fármacos que se liguem a formas transitórias menos evidentes — precisamos de uma maneira de gerar conjuntos inteiros de conformações plausíveis, e não apenas um único palpite.
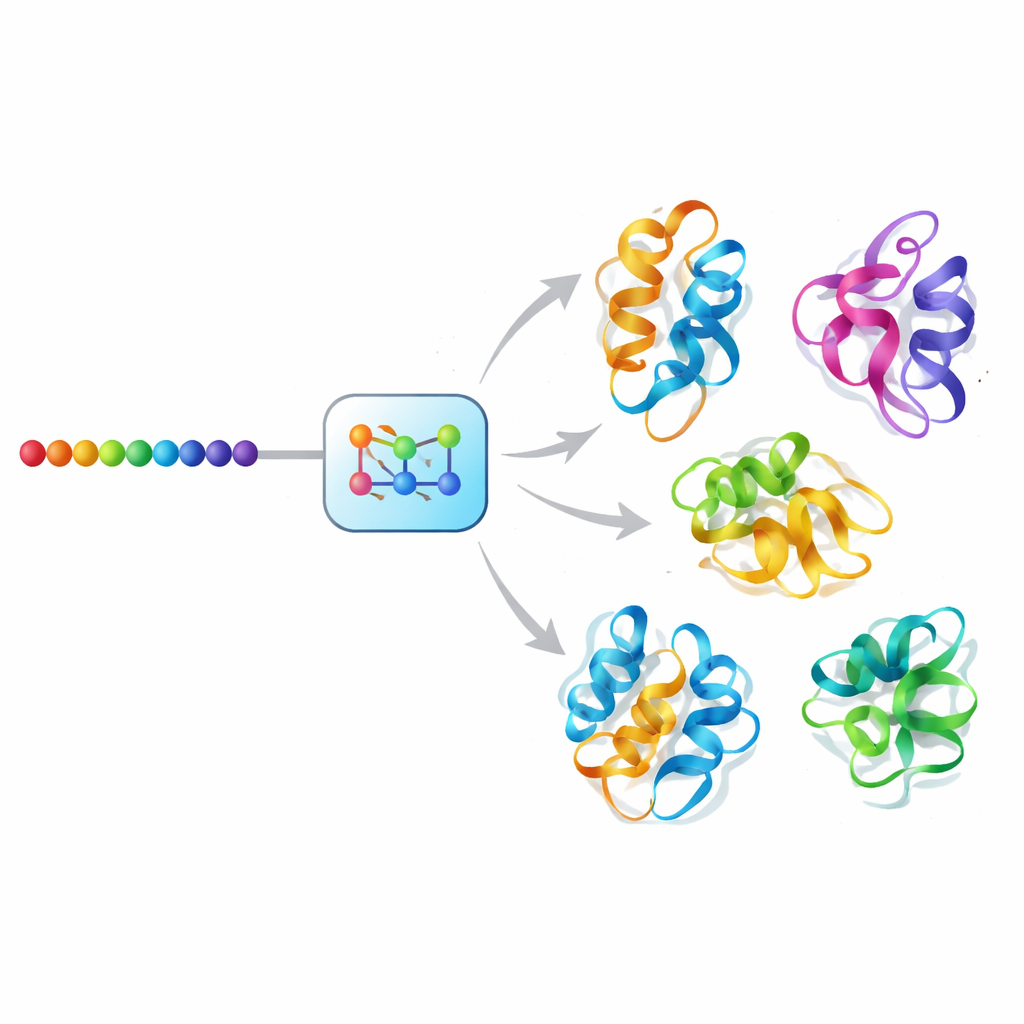
Uma abordagem de "difusão" por IA para o movimento protéico
O Mac-Diff enfrenta esse desafio usando um modelo generativo do tipo difusão, uma classe de IA que impulsionou avanços recentes em síntese de imagens. Em vez de remover ruído de fotografias, o Mac-Diff remove ruído de descrições geométricas abstratas dos esqueletos proteicos. O modelo representa uma proteína como uma grade de relações pareadas entre seus resíduos — distâncias e ângulos que são invariantes à rotação e translação da molécula como um todo. Em um passo direto, o sistema adiciona gradualmente ruído a esses padrões geométricos até que eles se assemelhem a estática aleatória. No passo reverso, ele aprende a remover o ruído passo a passo, guiado pela sequência de aminoácidos da proteína, até que geometria coerente compatível com 3D reapareça, podendo então ser convertida em modelos atômicos completos por softwares padrões de construção de estruturas.
Deixando a sequência falar com a estrutura localmente
Uma inovação chave está em como o Mac-Diff conecta uma sequência linear de resíduos aos seus vizinhos 3D. Permitir que cada resíduo “atenda” a todos os outros, como em modelos texto-para-imagem, borraria importantes restrições físicas. Em vez disso, os autores introduzem um mecanismo de atenção "sensível à localidade" que foca cada resíduo em uma pequena vizinhança provável de parceiros de interação. Para estimar essas vizinhanças, o Mac-Diff usa três ingredientes: um modelo de linguagem protéica chamado ESM-2, que codifica o contexto bioquímico de cada resíduo; um mapa de contatos que sugere quais pares de resíduos provavelmente estão próximos; e uma regra simples que favorece resíduos próximos ao longo da cadeia. Esses sinais são combinados de modo que, durante a desnoização, o modelo use preferencialmente informações de resíduos que sejam parceiros fisicamente plausíveis, afiando sua capacidade de reconstruir estruturas flexíveis e realistas.
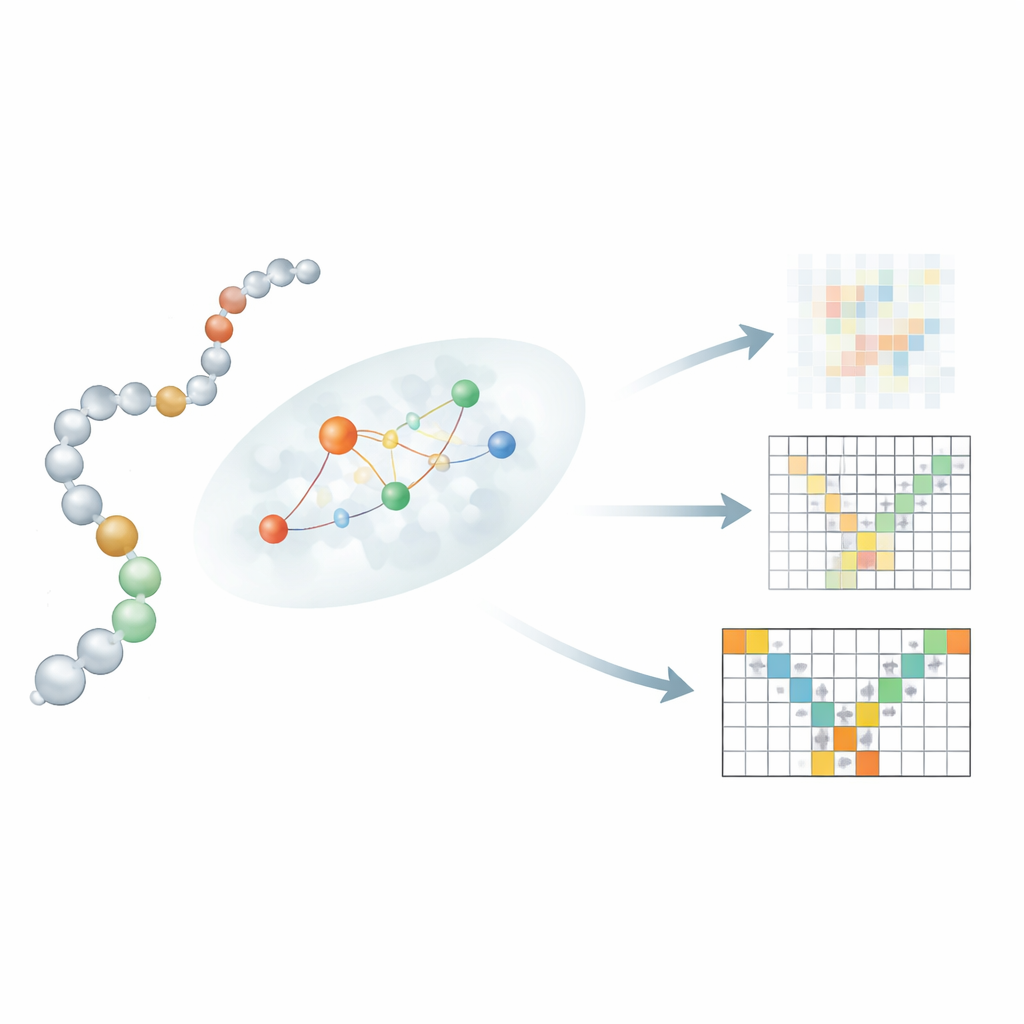
Testes contra simulações longas e proteínas que mudam de forma
Os pesquisadores testaram o Mac-Diff em duas frentes exigentes. Primeiro, verificaram se ele poderia reproduzir a ampla distribuição de formas observada em longas e cuidadosas simulações de dinâmica molecular de proteínas que dobram rapidamente e de uma proteína de referência clássica conhecida como BPTI. Em várias métricas que comparam propriedades estatísticas dos conjuntos gerados com os dados de simulação — como distribuições de distâncias internas e a compacidade geral — o Mac-Diff igualou ou superou métodos concorrentes de IA, ao mesmo tempo em que gerou uma variedade maior de conformações. Capturou a maioria dos principais estados “metastáveis” identificados nas simulações e reproduziu padrões de flexibilidade ao nível de resíduos com alta correlação, indicando que seus conjuntos refletem tanto dobras globais quanto oscilações locais de forma realista.
Revelando estados funcionais ocultos
Em segundo lugar, a equipe desafiou o Mac-Diff com proteínas conhecidas por adotarem formas muito diferentes enquanto desempenham suas funções, incluindo a enzima adenilato quinase, que alterna entre formas aberta e fechada durante o metabolismo energético, e um conjunto curado de 40 proteínas com duas conformações experimentalmente determinadas cada. O Mac-Diff gerou apenas 100 estruturas candidatas por proteína — bem menos que trajetórias típicas de simulação — e ainda assim recuperou a maioria dos estados conhecidos com bom acordo geométrico. Na adenilato quinase, por exemplo, produziu tanto conformações abertas quanto fechadas com alta semelhança às estruturas cristalográficas, enquanto vários métodos populares tenderam a favorecer apenas um estado. O modelo também rodou cerca de mil vezes mais rápido que simulações convencionais em hardware comparável, tornando a exploração sistemática da diversidade de formas muito mais prática.
O que isso significa para a biologia e a medicina
Em termos práticos, o Mac-Diff transforma a sequência de uma proteína em uma galeria de poses plausíveis em vez de um único retrato, e faz isso com sensibilidade sobre quais partes tendem a empurrar ou agarrar umas às outras em 3D. Ao amostrar esses conjuntos com precisão e eficiência, o método oferece uma maneira de sondar como mudanças sutis de forma sustentam função, identificar conformações raras mas importantes e buscar bolsões de ligação a fármacos que aparecem apenas em estados transitórios. Embora ainda não capture os filmes ordenados no tempo que as simulações fornecem, o Mac-Diff aproxima a paisagem dinâmica das proteínas para muitos mais sistemas, prometendo novos insights em biologia estrutural, desenho de fármacos e engenharia de proteínas.
Citação: Wang, B., Wang, C., Chen, J. et al. Conditional diffusion with locality-aware modal alignment for generating diverse protein conformational ensembles. Nat Mach Intell 8, 415–434 (2026). https://doi.org/10.1038/s42256-026-01198-9
Palavras-chave: dinâmica de proteínas, modelos de difusão, conjuntos conformacionais, proteínas alostéricas, descoberta de fármacos