Clear Sky Science · pt
Robustez sem precedentes de modelos de energia atômica informados pela física em e além da temperatura ambiente
Por que isso importa para a química do dia a dia
Simulações por computador são as ferramentas de trabalho da química e da ciência dos materiais modernas. Elas permitem aos pesquisadores observar moléculas torcerem, vibrarem e colidirem in silico em vez de em experimentos dispendiosos e demorados. Mas quando essas simulações dependem de aprendizado de máquina, elas podem de repente "explodir", produzindo formas moleculares impossíveis — especialmente em temperaturas mais altas. Este estudo introduz um novo tipo de modelo de aprendizado de máquina informado pela física que consegue executar tais simulações por tempos muito longos, em temperaturas de até 1000 kelvin, sem se desfazer.
De atalhos engenhosos a simulações frágeis
A química quântica tradicional calcula energias moleculares com alta precisão, mas é dolorosamente lenta. Campos de força mais simples são rápidos, porém frequentemente aproximados. Potenciais aprendidos por máquina buscam combinar o melhor dos dois mundos: eles aprendem um atalho da geometria molecular até energia e forças, e então usam esse atalho para conduzir a dinâmica molecular. No papel, muitos desses modelos parecem excelentes, exibindo erros médios mínimos em conjuntos de teste padrão. Na prática, esses números podem ser enganosos. Quando as moléculas exploram novas formas durante uma simulação — especialmente em temperaturas mais altas — muitos modelos são levados para fora do intervalo de estruturas em que foram treinados. Em vez de direcionar suavemente as moléculas de volta a formas realistas, eles podem prever forças que esticam ou esmagam ligações até que todo o sistema se torne não físico e a simulação trave.
Construindo modelos a partir de blocos quânticos
Os autores enfrentam essa fragilidade mudando o que o modelo aprende e como ele é guiado por conhecimentos físicos prévios. Eles usam uma estrutura chamada FFLUX, que se baseia na abordagem Interacting Quantum Atoms (IQA). Na IQA, uma molécula é dividida em "átomos topológicos" cujas energias individuais são determinadas diretamente da mecânica quântica. Essas energias atômicas têm significado físico e se somam à energia total da molécula. Em vez de aprender energias de sítio arbitrárias, os novos modelos por processo gaussiano aprendem essas energias atômicas enraizadas na teoria quântica, fornecendo uma âncora física profunda para cada previsão. Quatro moléculas orgânicas flexíveis — glicina e serina com terminal protegido por peptídeo, malondialdeído e aspirina — servem como corpos de prova desafiadores devido aos seus muitos movimentos internos e à dificuldade conhecida para campos de força aprendidos por máquina existentes.
Ensinar o modelo a esperar problemas
Uma inovação chave está em como o processo gaussiano é configurado antes mesmo de ver dados: sua "função média", que codifica o que o modelo assume em regiões pouco conhecidas. A maioria dos trabalhos anteriores simplesmente define essa média como zero, praticamente fingindo que o modelo não tem expectativas prévias. Os autores, em vez disso, deslocam deliberadamente essa média em direção a estados atômicos de energia mais alta, mantendo-a fisicamente plausível. Essa escolha de projeto significa que quando o modelo é forçado a extrapolar — por exemplo, quando ligações estão temporariamente muito esticadas — ele prefere naturalmente previsões que penalizam distorções extremas. Em testes extensivos, versões do modelo que diferiam apenas nesse prior se comportaram de maneira muito distinta. Modelos com médias convencionais ou de baixa energia frequentemente sobreviviam menos de um picosegundo antes de a molécula explodir ou implodir. Em contraste, a melhor média de alta energia (apelidada MF5) produziu simulações que permaneceram estáveis durante toda a janela de teste de nanosegundos, em temperaturas de 300 a 1000 kelvin, para todas as quatro moléculas.
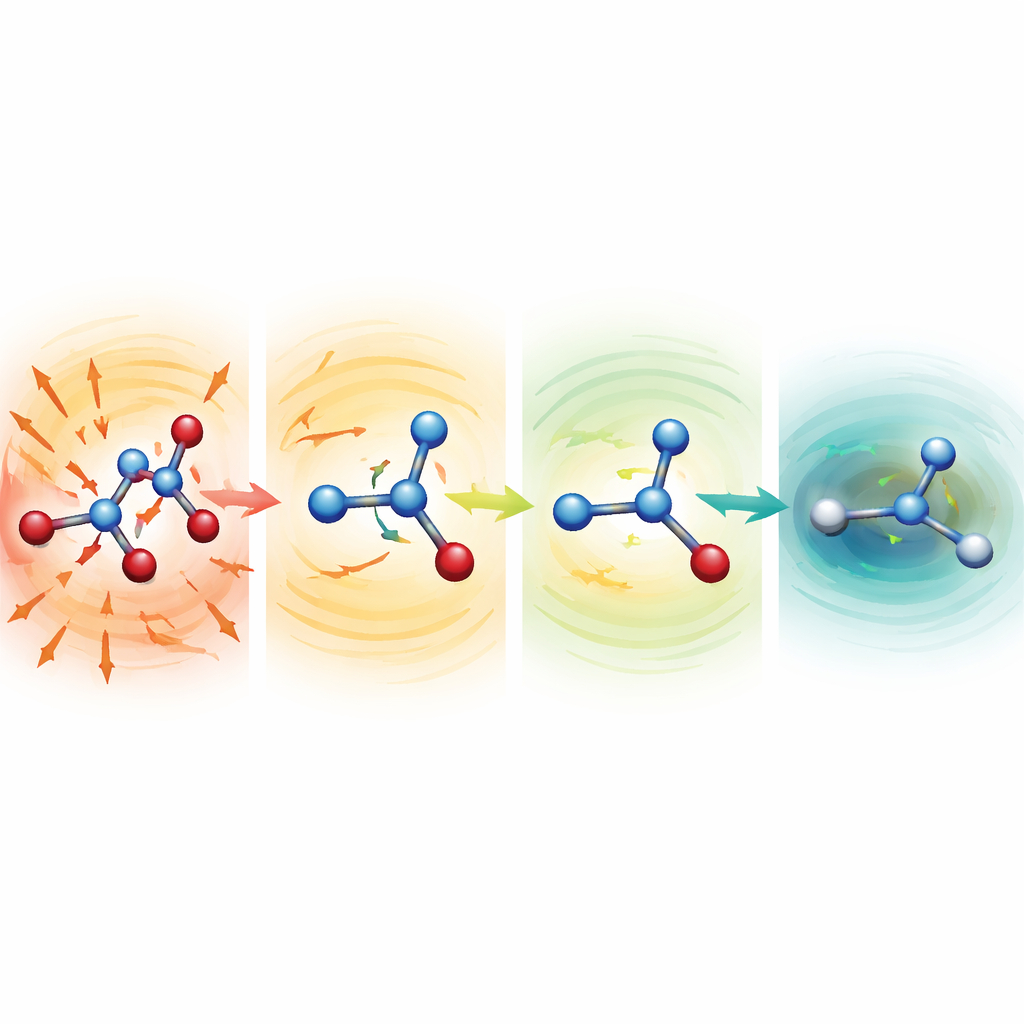
Ver moléculas distorcidas se repararem
Para investigar por que os modelos robustos funcionam tão bem, os pesquisadores iniciaram simulações a partir de estruturas deliberadamente danificadas com ligações severamente esticadas ou comprimidas. Para serina, aspirina e malondialdeído, esses pontos iniciais situavam-se a centenas até mais de mil quilocalorias por mol acima de uma estrutura normal — configurações que normalmente seriam desastrosas. Com funções médias mais fracas, as moléculas rapidamente se despedaçaram. Com a configuração MF5, porém, as forças previstas apontaram imediatamente em direções que encurtaram ligações alongadas e alongaram as comprimidas. Em dezenas a centenas de passos de tempo, as moléculas relaxaram para formas realistas e então continuaram a evoluir de maneira estável. A equipe também mostrou que, sem nunca terem sido treinados em forças, esses mesmos modelos podem orientar otimizações de geometria do dipeptídeo de alanina, reproduzindo conformações de baixa energia conhecidas e energias relativas dentro de algumas décimas de quilocaloria por mol, porém com custo cerca de 200 vezes menor do que cálculos quânticos completos.
Simulações longas e quentes em hardware comum
Robustez não é apenas sobreviver a um começo difícil; é resistir por milhões ou bilhões de passos de tempo. Os autores levaram seus melhores modelos adiante executando 50 simulações independentes a 500 kelvin, cada uma por 10 nanosegundos, nas quatro moléculas de teste. Nenhuma dessas execuções travou, resultando em um tempo de simulação combinado de meio microssegundo — incomum para campos de força aprendidos por máquina de ponta. Ainda mais impressionante, as simulações rodaram de forma eficiente em CPUs padrão, passo a passo rivalizando ou superando alguns potenciais por redes neurais proeminentes que exigem GPUs potentes. Ao longo das execuções, as moléculas exploraram conjuntos ricos de formas e estados metastáveis, mostrando que a robustez não foi obtida por congelar artificialmente o movimento nem por impor estruturas rígidas.
O que isso significa para a modelagem molecular futura
Para não especialistas, a mensagem chave é que nem todos os modelos de aprendizado de máquina com baixo erro são confiáveis quando empurrados ao extremo. Ao fundamentar seus modelos em energias atômicas derivadas da teoria quântica e ajustar cuidadosamente as expectativas embutidas do modelo em direção a estados de alta energia, os autores criaram uma família de potenciais que produzem naturalmente "forças restauradoras" — o equivalente molecular a um cinto de segurança — que mantêm as simulações físicas mesmo em altas temperaturas e a partir de pontos de partida distorcidos. Essa abordagem promete simulações mais longas e mais confiáveis de moléculas complexas, e aponta para extensões futuras onde modelos informados pela física semelhantes lidem com fases condensadas e interações sutis como dispersão, tudo isso mantendo viabilidade computacional.
Citação: Isamura, B.K., Aten, O., Nosratjoo, M. et al. Unprecedented robustness of physics-informed atomic energy models at and beyond room temperature. Commun Chem 9, 138 (2026). https://doi.org/10.1038/s42004-026-01965-0
Palavras-chave: campos de força por aprendizado de máquina, robustez em dinâmica molecular, potenciais por processo gaussiano, modelagem informada pela física, energias atômicas quânticas