Clear Sky Science · pt
Aprimorando o AlphaFold com observações de crio‑EM limitadas
Por que é tão difícil ver as formas das proteínas
Proteínas são pequenas máquinas moleculares que impulsionam quase todos os processos em nossos corpos, desde a produção de energia até a transmissão de sinais nervosos. Para entender como elas funcionam — e como medicamentos podem controlá‑las — os cientistas precisam conhecer suas formas tridimensionais precisas. Duas ferramentas poderosas surgiram para essa tarefa: a crio–microscopia eletrônica (crio‑EM), que captura muitas imagens borradas de proteínas congeladas, e o AlphaFold, um sistema de inteligência artificial que prevê estruturas de proteínas a partir de suas sequências. Mas em muitos experimentos reais os dados de crio‑EM são incompletos, e as previsões do AlphaFold nem sempre correspondem à realidade. Este artigo apresenta o CoCoFold, um método que ensina o AlphaFold a ouvir diretamente dados difíceis de crio‑EM e a melhorar suas previsões em conformidade.
Quando a câmera enxerga de menos
A crio‑EM funciona ao congelar rapidamente proteínas e registrar um número enorme de partículas individuais de muitos ângulos, combinando essas imagens em um mapa 3D. Na prática, porém, os pesquisadores muitas vezes não dispõem de imagens boas o suficiente. Às vezes a proteína aparece brevemente em um estado de alta energia, então muito poucas partículas são capturadas. Em outros casos, as proteínas preferem certas orientações sobre a superfície do gelo, fazendo com que muitos ângulos de visão faltem. Ambos os problemas produzem mapas borrados e incompletos, difíceis de traduzir em modelos atômicos confiáveis. Softwares existentes podem encaixar as estruturas previstas pelo AlphaFold nesses mapas, mas seu sucesso depende fortemente de dispor de dados nítidos e de alta resolução desde o início.
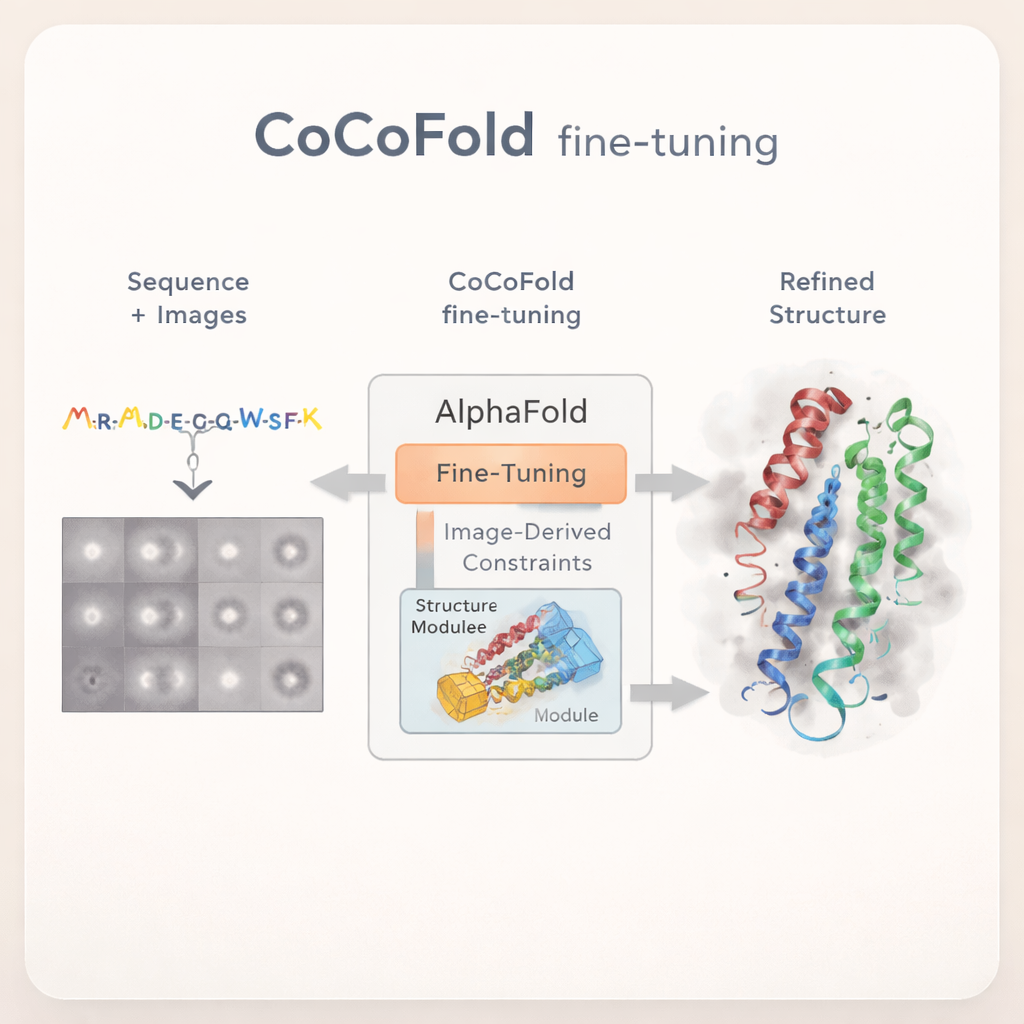
Ensinando o AlphaFold a aprender com imagens brutas
O CoCoFold adota uma abordagem diferente: em vez de confiar em um mapa 3D de crio‑EM totalmente reconstruído, ele usa diretamente as imagens 2D brutas das partículas para ajustar finamente o AlphaFold. O método parte de uma previsão AlphaFold‑Multimer e mantém a maior parte da rede original congelada, preservando seu amplo conhecimento sobre dobramento de proteínas. Apenas a parte final de construção da estrutura é permitida a mudar. Um “adaptador” leve é adicionado para alimentar informações derivadas das imagens de crio‑EM nesse módulo de estrutura, empurrando suavemente o modelo em direção a formas compatíveis com os dados experimentais enquanto evita desvios extremos da física conhecida das proteínas.
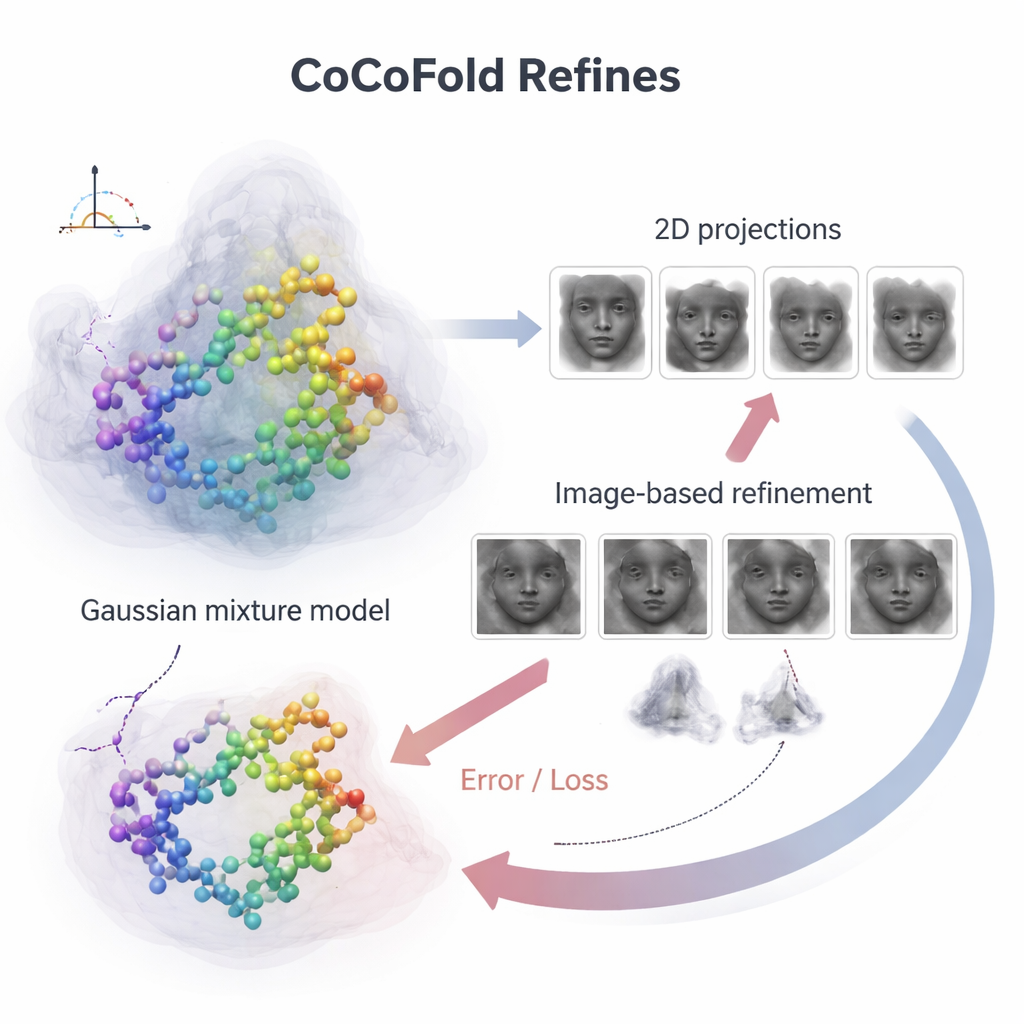
Transformando imagens em feedback estrutural
Para conectar átomos individuais da proteína às imagens ruidosas do microscópio, o CoCoFold constrói uma representação suave e flexível da estrutura prevista usando blobs tridimensionais sobrepostos, conhecidos como mistura Gaussianas. A partir dessa representação ele simula como a proteína apareceria no microscópio nas mesmas direções de visualização e condições de imagem do experimento real. Esses instantâneos simulados são então comparados às partículas reais de crio‑EM, anel por anel no domínio da frequência, para avaliar o grau de correspondência. Qualquer descompasso torna‑se um sinal de feedback que volta através da rede, ajustando levemente tanto o modelo da proteína quanto a representação de densidade. Após o treinamento, o modelo atômico é ainda refinado com um passo baseado em física para remover conflitos geométricos locais.
Mantendo precisão quando os dados são escassos ou tendenciosos
Os autores testaram o CoCoFold em vários conjuntos de dados experimentais e simulados projetados para imitar os dois problemas principais na crio‑EM: poucas partículas e grandes lacunas em ângulos de visão. Nessas condições difíceis, ferramentas padrão — incluindo outros métodos de aprendizado profundo que dependem de mapas reconstruídos — tendiam a perder regiões da proteína, deslocar hélices ou perder detalhes finos conforme os mapas se tornavam mais borrados. O CoCoFold, por outro lado, produziu consistentemente modelos que correspondiam mais de perto e de forma mais completa às estruturas de referência conhecidas. Seus erros permaneceram pequenos mesmo quando o número de partículas foi drasticamente reduzido ou quando grandes cones de direções de visualização estavam ausentes, sugerindo que aprender diretamente com as imagens brutas preserva informações cruciais que abordagens baseadas em mapa descartam.
O que isso significa para a biologia estrutural futura
Para não‑especialistas, a mensagem principal é que o CoCoFold age como um tradutor entre previsões poderosas de IA e dados experimentais imperfeitos. Em vez de confiar apenas no AlphaFold ou apenas na crio‑EM, ele permite que ambos se informem mutuamente, especialmente nos regimes difíceis em que os experimentos fornecem apenas uma visão parcial. Em casos simples, com dados abundantes e de alta qualidade, ferramentas baseadas em mapas ainda funcionam extremamente bem. Mas quando partículas são raras ou orientações estão faltando — situações comuns ao perseguir estados proteicos efêmeros ou frágeis — o CoCoFold oferece uma maneira de recuperar modelos atômicos confiáveis a partir de informações que, de outra forma, seriam desperdiçadas.
Citação: Liao, J., Zheng, D., Zhang, H. et al. Fine-tuning AlphaFold with limited cryo-EM observations. Commun Chem 9, 95 (2026). https://doi.org/10.1038/s42004-026-01899-7
Palavras-chave: crio‑EM, AlphaFold, estrutura de proteínas, aprendizado profundo, biologia estrutural