Clear Sky Science · pt
Falha energética mitocondrial subjaz à neuropatia sensorial relacionada ao FLVCR1
Quando os nervos da dor ficam sem energia
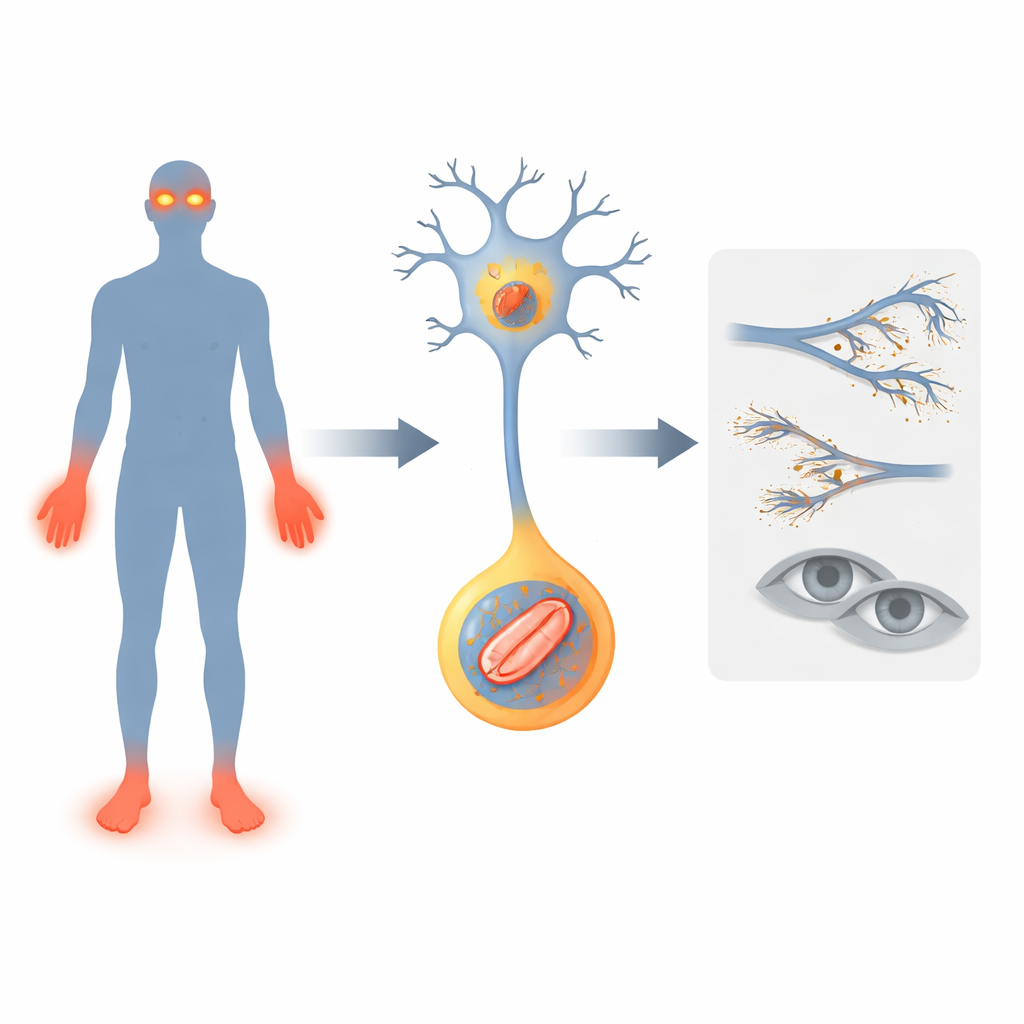
Algumas pessoas nascem quase incapazes de sentir dor. À primeira vista isso pode soar como uma vantagem, mas rapidamente se torna uma maldição: sem a dor como sinal de alerta, acumulam-se queimaduras, fraturas, infecções e até cegueira. Este estudo investiga uma forma rara desses distúrbios de perda da dor e revela um culpado surpreendente: minúsculas usinas de energia dentro das células nervosas cuja produção energética se altera de forma grave.
Um gene que silencia os alarmes
Os pesquisadores concentram-se em um gene chamado FLVCR1, já associado a condições nervosas raras em que as pessoas perdem a sensação de dor, desenvolvem marcha instável e, às vezes, perda progressiva da visão. Eles descrevem dois novos pacientes com alterações no FLVCR1. Ambas as crianças apresentaram problemas precoces: atraso nas aquisições motoras, quedas frequentes, infecções profundas e mutilação de dedos das mãos e dos pés porque as lesões passavam despercebidas. Uma delas também desenvolveu uma doença degenerativa ocular chamada retinite pigmentosa, levando à cegueira noturna. Esses casos ampliam o quadro de como defeitos em FLVCR1 podem se manifestar em humanos e reforçam a ideia de que esse gene é vital para manter vivos os neurônios sensoriais de dor e as células sensíveis à luz da retina.
Modelando a doença em peixinhos
Para explorar como o FLVCR1 afeta os nervos sensoriais em desenvolvimento, a equipe recorreu ao peixe-zebra, cujos embriões transparentes permitem a visualização direta das células nervosas. Eles reduziram os níveis da versão do gene no peixe, flvcr1a, usando ferramentas genéticas. Peixes com flvcr1a reduzido tinham menos gânglios da raiz dorsal, aglomerados de neurônios que detectam toque e dor ao longo da coluna. Comportamentalmente, esses peixes moviam-se menos espontaneamente e nadavam apenas curtas distâncias quando a cauda era tocada suavemente, sugerindo uma resposta sensorial diminuída. Como modelos de camundongo anteriores morriam cedo demais para permitir a análise dos nervos sensoriais, esses peixes-zebra fornecem o primeiro sistema vivo no qual os defeitos nervosos e comportamentais relacionados ao FLVCR1 podem ser acompanhados em detalhe.
Três vias afetadas convergem nas usinas celulares
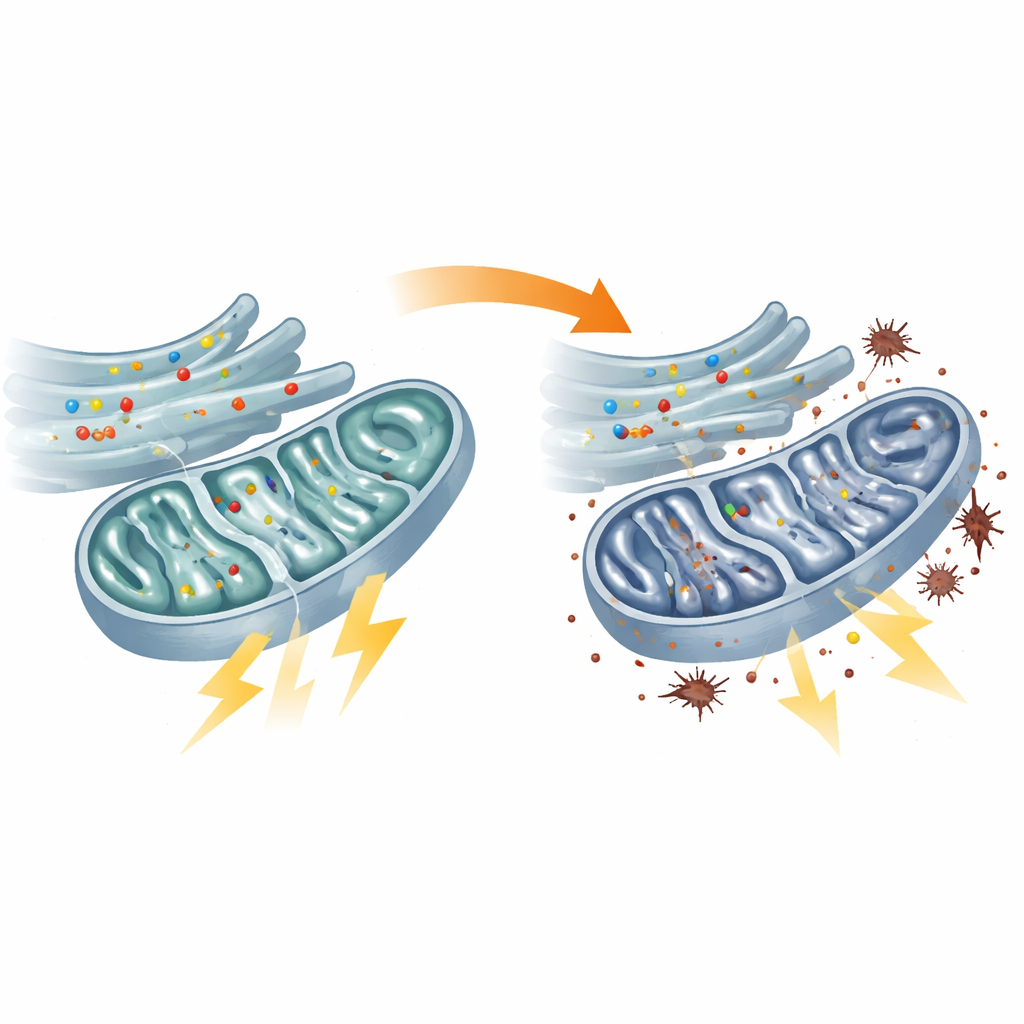
O FLVCR1 localiza-se nas membranas celulares e gerencia várias substâncias-chave. Trabalhos anteriores sugeriram papéis no manejo de colina (um bloco de construção dos lipídios de membrana), heme (o pigmento contendo ferro que alimenta muitas enzimas) e no fluxo de cálcio entre compartimentos celulares. Os cientistas coletaram células da pele (fibroblastos) de quatro pacientes portadores de diferentes mutações em FLVCR1 e as compararam com células de pessoas saudáveis e de portadores assintomáticos. Eles descobriram que as células dos pacientes tinham níveis menores de colina e membranas celulares mais fluidas, alterações que poderiam perturbar o ambiente lipídico delicado exigido pelas mitocôndrias, os organelos geradores de energia. Também verificaram que uma enzima crucial para produzir heme dentro das mitocôndrias, ALAS1, estava menos ativa, embora o conteúdo total de heme parecesse quase normal. Ao mesmo tempo, os sítios de contato físico entre o retículo endoplasmático e as mitocôndrias — por onde o cálcio normalmente entra nas mitocôndrias — eram mais curtos e menos frequentes, e a entrada de cálcio nas mitocôndrias estava reduzida. Três problemas — falta de colina, produção de heme lenta e transferência de cálcio enfraquecida — apontavam todos para um desempenho mitocondrial comprometido.
Mitocôndrias famintas e sistemas de reserva sobrecarregados
Testes diretos do metabolismo energético confirmaram que as mitocôndrias nos fibroblastos dos pacientes estavam com desempenho reduzido. O centro de processamento de combustíveis conhecido como ciclo do TCA funcionou mais lentamente, várias de suas enzimas-chave estavam menos ativas e a cadeia de reações que normalmente converte combustível em ATP, a moeda energética da célula, estava prejudicada. Como resultado, os níveis de ATP dentro das mitocôndrias caíram. As células tentaram compensar aumentando a glicólise, uma via menos eficiente de queima de açúcar fora das mitocôndrias. Essa mudança de estratégia energética teve um custo: elétrons vazaram da maquinaria mitocondrial estressada e desencadearam níveis maiores de peroxidação lipídica, uma forma de dano oxidativo às membranas celulares. Defeitos semelhantes foram observados em peixes-zebra com flvcr1a reduzido, ligando diretamente a falha mitocondrial ao modelo animal de neuropatia sensorial.
Pistas para tratamentos futuros ao reforçar a energia celular
De forma encorajadora, alguns desses defeitos puderam ser amenizados em laboratório. Quando a equipe aumentou artificialmente a entrada de cálcio nas mitocôndrias ao superexpressar uma proteína canal chamada MCU nas células de pacientes, a produção de energia recuperou-se e sinais de dano oxidativo diminuíram. Fornecer às células um precursor do heme, o ácido 5-aminolevulínico (ALA), também melhorou a atividade do ciclo do TCA, a função da cadeia respiratória e os níveis de ATP, embora exposições prolongadas a ALA tenham sido prejudiciais em estudos anteriores. Colina suplementar normalizou a fluidez da membrana e ajudou a reduzir o dano lipídico, mas trouxe ganhos modestos e de curto prazo na produção de energia. Esses experimentos de resgate sugerem que nenhuma via isolada é a única responsável; em vez disso, uma rede de alterações no manejo da colina, do heme e do cálcio empurra as mitocôndrias para um desempenho cronicamente insuficiente.
Por que essas descobertas importam para os pacientes
Ao traçar as consequências das mutações em FLVCR1 desde as moléculas até as células e organismos inteiros, este trabalho propõe que a falha de energia nas mitocôndrias é uma força motriz por trás desta forma de neuropatia por perda de dor e seus problemas visuais associados. Nervos sensoriais e fotorreceptores têm necessidades energéticas incomumente altas porque mantêm axônios longos ou renovam continuamente estruturas sensíveis à luz, tornando-os especialmente vulneráveis quando a produção mitocondrial falha. O modelo com peixe-zebra e as células derivadas de pacientes agora oferecem campos de teste práticos para terapias que fortaleçam o metabolismo mitocondrial. Embora tratamentos como suplementação de colina, aumento controlado do heme ou fármacos que aumentem a captação de cálcio mitocondrial exijam avaliação cuidadosa em modelos animais e ensaios clínicos, a mensagem central é clara: restaurar a fonte de energia de neurônios frágeis pode um dia ajudar a proteger pessoas nascidas sem o sinal de alerta mais importante da natureza — a dor.
Citação: Bertino, F., Zanin Venturini, D.I., Grasso, E. et al. Mitochondrial energetic failure underlies FLVCR1-related sensory neuropathy. Commun Biol 9, 429 (2026). https://doi.org/10.1038/s42003-026-09691-y
Palavras-chave: neuropatia sensorial, disfunção mitocondrial, FLVCR1, insensibilidade à dor, metabolismo energético nervoso