Clear Sky Science · pt
Um guia prático das tecnologias de sequenciamento de RNA de célula única direcionado
Por que analisar células individuais importa
Cada célula do seu corpo carrega o mesmo DNA, mas diferentes células se comportam de maneiras muito distintas. Elas fazem isso ativando ou desativando genes específicos e editando moléculas de RNA de formas sutis. O sequenciamento moderno de RNA de célula única pode ler quais RNAs estão presentes em milhares de células ao mesmo tempo, mas atualmente perde a maior parte da mensagem. Esta revisão explica onde as técnicas atuais perdem informação e como novos métodos “direcionados” estão sendo desenvolvidos para focalizar as partes mais importantes das moléculas de RNA para pesquisa, diagnóstico e desenho de tratamentos.
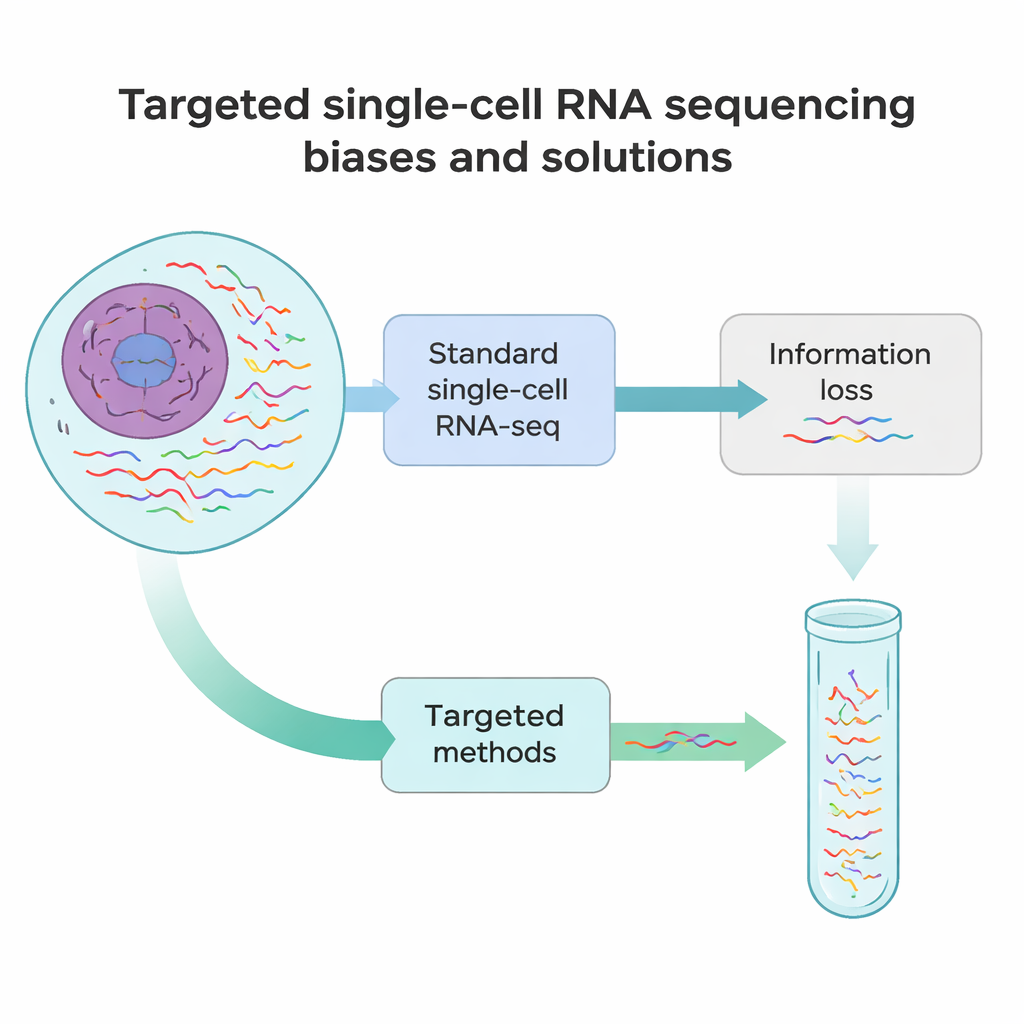
Onde os métodos atuais ficam aquém
O sequenciamento padrão de RNA de célula única funciona um pouco como tirar uma foto rápida de cada mensagem na célula em vez de um filme de longa duração. Na maioria dos experimentos, apenas cerca de 10–40% de todos os RNAs em uma célula são detectados, e apenas seu início ou fim é lido. Isso significa que muitos RNAs raros, porém importantes — como marcadores que definem a identidade celular ou variantes de genes que carregam mutações causadoras de doença — são facilmente perdidos. Além disso, várias etapas técnicas, desde dissociar tecidos em células únicas até converter RNA em DNA e amplificá-lo, introduzem vieses sistemáticos. Alguns RNAs são interrompidos cedo, alguns ficam superrepresentados e outros desaparecem completamente dos dados.
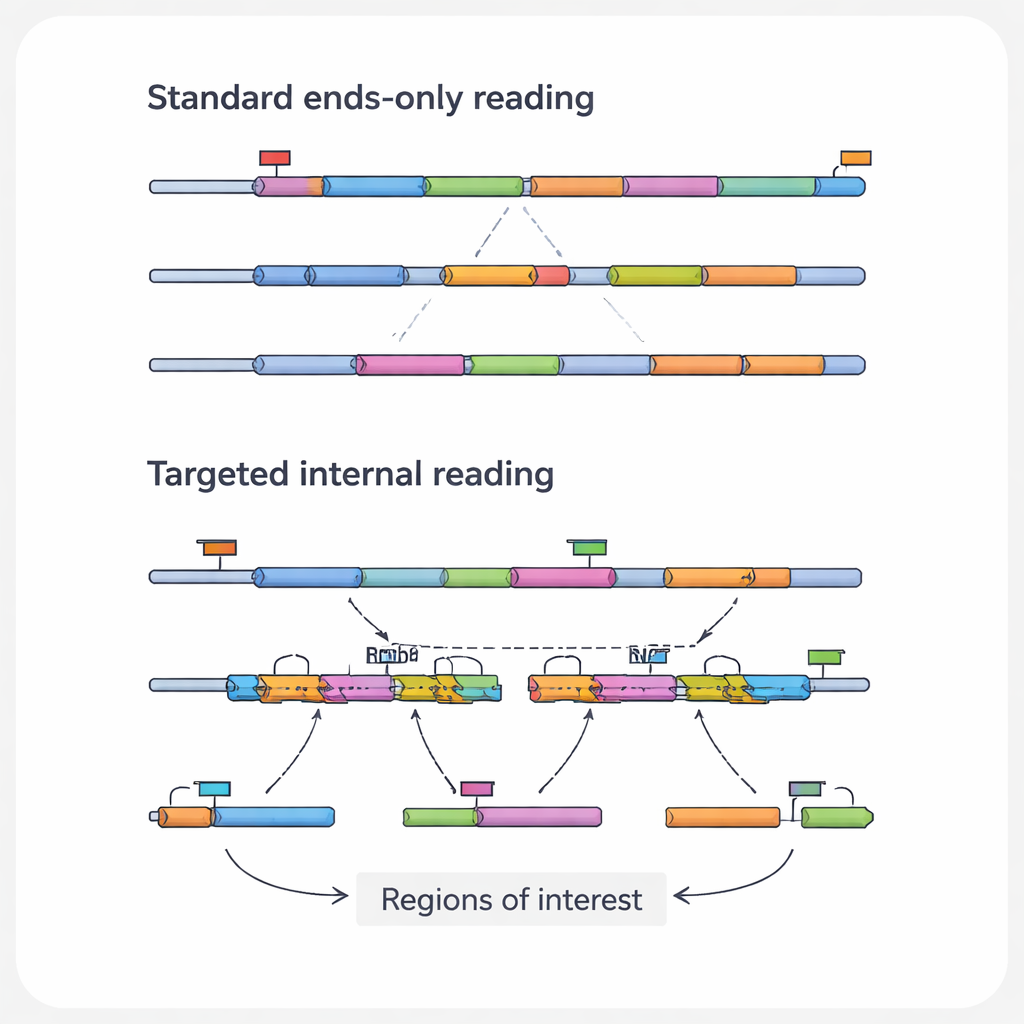
Por que detalhes internos do RNA importam
A informação mais relevante medicamente em uma molécula de RNA frequentemente está em suas regiões internas, não nas extremidades que os métodos padrão conseguem ver. Essas seções internas podem conter mutações pontuais que impulsionam o câncer, pontos de fusão onde dois genes foram anormalmente juntados, ou junções de splicing que criam variantes proteicas diferentes a partir do mesmo gene. Podem também registrar as marcas de ferramentas de edição genética, como CRISPR. Os autores chamam essas características específicas de “regiões de interesse”, e os RNAs que as carregam de “transcritos de interesse”. Como as plataformas de alto rendimento comuns leem principalmente as pontas dos RNAs, elas rotineiramente negligenciam esses detalhes cruciais, especialmente em transcritos longos ou de baixa abundância.
Novas maneiras de apontar o holofote
Para superar esses pontos cegos, pesquisadores desenvolveram uma família de abordagens de sequenciamento de RNA de célula única direcionado. Em vez de tentar ler todos os RNAs igualmente, esses métodos enriquecem deliberadamente transcritos ou regiões selecionadas. Algumas estratégias redesenham as esferas de captura para que se prendam a sequências internas de RNA em vez de apenas à cauda, puxando mensagens escolhidas para a biblioteca desde a primeira etapa. Outras adicionam primers personalizados que iniciam a cópia em um ponto interno, ou etapas extras de PCR que amplificam especificamente uma lista curta de genes a partir de uma biblioteca existente. Outro conjunto usa sondas de DNA que hibridizam com RNAs-alvo ou suas cópias e então os pescam, frequentemente com etiquetas químicas simples. Cada categoria troca sensibilidade, número de células, número de alvos e custo, mas todas têm o mesmo objetivo: recuperar mais detalhes relevantes a partir das mesmas leituras de sequenciamento ou de menos leituras.
Aplicações de vírus a tumores
Esses métodos direcionados já estão remodelando várias áreas da biologia e da medicina. Em infecções, eles podem finalmente capturar RNAs virais ou bacterianos que não possuem as caudas poli(A) que os protocolos padrão esperam, revelando em quais células hospedeiras eles habitam e como alteram a atividade gênica do hospedeiro. No câncer, o sequenciamento de célula única direcionado pode identificar quais tipos celulares carregam mutações específicas ou genes de fusão e ligar isso a programas gênicos alterados, ajudando a explicar por que algumas células se tornam resistentes à terapia. Outros métodos focam em splicing alternativo, descobrindo quais tipos celulares usam quais isoformas, ou em populações celulares raras e marcadores sutis que, de outra forma, ficariam abaixo do limiar de detecção. Em triagens CRISPR em pool, a captura aprimorada de RNAs guias permite que cientistas associem cada perturbação genética à sua resposta celular precisa.
Escolhendo a ferramenta certa e o que vem a seguir
Como agora existe uma caixa de ferramentas lotada de abordagens direcionadas, os autores propõem uma árvore de decisão para ajudar pesquisadores a escolher um método. Perguntas-chave incluem se é necessário o perfil completo do transcriptoma, quantos genes ou regiões devem ser alvo, quão distantes essas regiões estão das extremidades do RNA e quantas células podem ser processadas. Olhando à frente, eles argumentam que os maiores ganhos virão da melhoria das etapas iniciais de captura, da expansão de estratégias inteligentes baseadas em sondas e da combinação de direcionamento com plataformas emergentes de sequenciamento de leitura longa e sequenciamento direto de RNA. Até que se torne prático ler todo RNA em cada célula de ponta a ponta, o sequenciamento de RNA de célula única direcionado continuará essencial para ver as partes da mensagem celular que mais importam para a biologia e a doença.
Citação: Moro, G., Brunner, E. & Basler, K. A practical guide to targeted single-cell RNA sequencing technologies. Commun Biol 9, 250 (2026). https://doi.org/10.1038/s42003-026-09675-y
Palavras-chave: sequenciamento de RNA de célula única, sequenciamento direcionado, transcriptômica, mutações cancerígenas, transcriptômica espacial