Clear Sky Science · pt
A desmetilase de histona KDM7A regula negativamente a polarização de macrófagos fibróticos e a progressão da fibrose pulmonar
Por que o enrijecimento dos pulmões importa para todos
Quando os pulmões desenvolvem cicatrizes persistentes, respirar torna-se uma luta diária. Essa condição, conhecida como fibrose pulmonar, afeta milhões e atualmente não tem cura — há apenas medicamentos que retardam o dano. Neste estudo, os pesquisadores descobrem um “freio” molecular até então escondido dentro de células imunológicas chamadas macrófagos que ajuda a conter a formação de cicatrizes pulmonares. Entender esse freio pode abrir caminho para novos tratamentos não apenas para fibrose pulmonar, mas potencialmente para outras doenças em que cicatrizes prejudiciais e inflamação desenfreada andam juntas.
Uma história de células imunológicas que mudam de forma
Macrófagos são células de linha de frente do sistema imunológico que patrulham tecidos, removem detritos e ajudam a reparar danos. Mas eles também mudam de forma: em algumas situações tornam-se combatentes pró-inflamatórios, enquanto em outras se transformam em curadores de feridas que podem estimular a formação de cicatrizes. Um tipo específico que promove cicatrizes, chamado macrófagos profibróticos (Fib-Mac), está fortemente associado à fibrose pulmonar. Essas células produzem moléculas que ativam fibroblastos, que então depositam excesso de colágeno e outros componentes de matriz, endurecendo gradualmente o pulmão. Os autores queriam saber como as “configurações” genéticas dentro dos macrófagos decidem se eles se tornam essas perigosas células Fib-Mac ou permanecem em estados mais equilibrados e protetores.

Um freio epigenético escondido no genoma
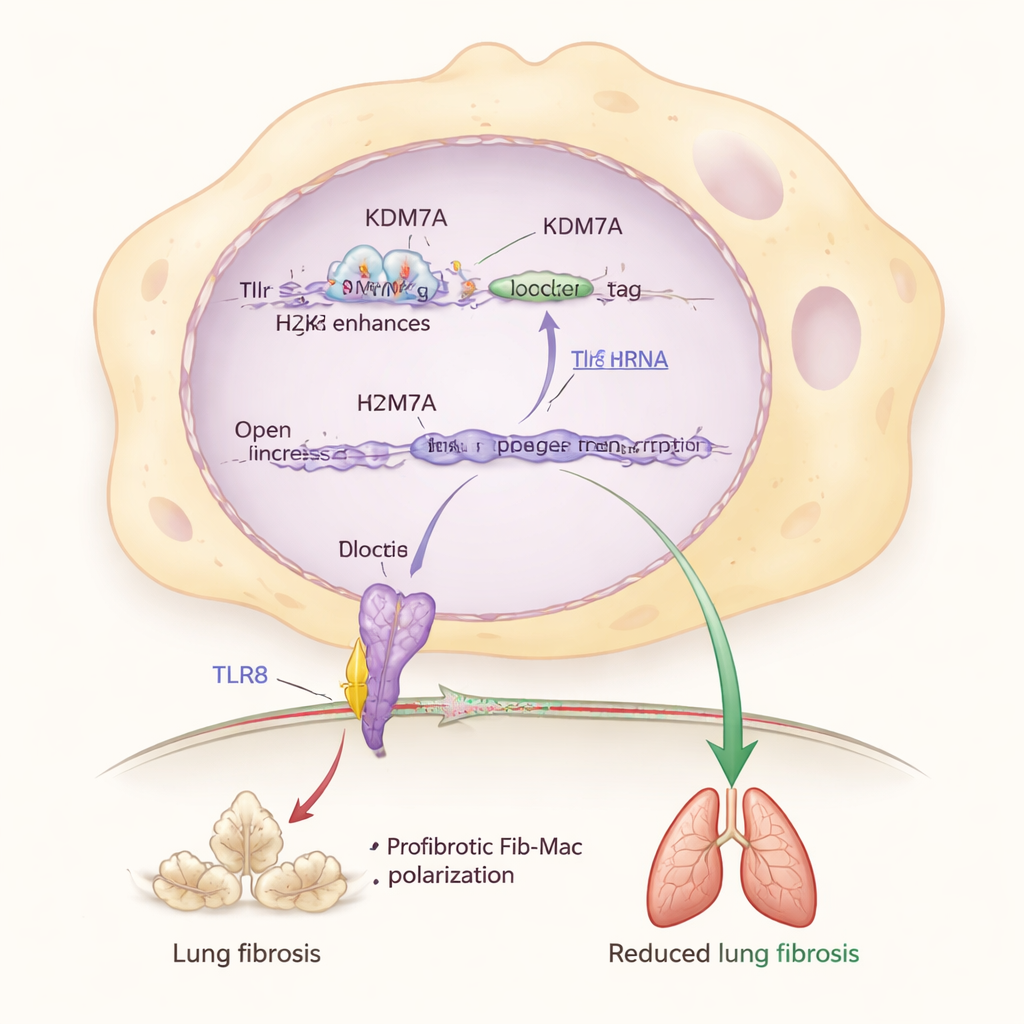
A equipe começou vasculhando centenas de reguladores epigenéticos conhecidos — proteínas que ajustam quão compactado o DNA está e quais genes são ativados ou silenciados. Usando sequenciamento de RNA em macrófagos humanos e de camundongo, eles descobriram que uma enzima chamada KDM7A era fortemente ativada quando os macrófagos eram direcionados para um estado fibrótico, de cicatrização. KDM7A é uma “desmetilase de histona”: remove certas marcas químicas das proteínas histonas em torno das quais o DNA se enrola. Esse padrão sugeriu que KDM7A poderia agir como um freio de feedback embutido, ativado precisamente quando os macrófagos começam a deslizar em direção a uma identidade que promove cicatrizes.
Para testar isso, os pesquisadores usaram camundongos que não têm o gene Kdm7a e provocaram lesão pulmonar com o medicamento quimioterápico bleomicina, um modelo padrão de fibrose pulmonar. No início após a lesão, o tecido pulmonar parecia similar em animais normais e deficientes em Kdm7a. Mas em três semanas, os camundongos sem Kdm7a apresentaram cicatrizes muito mais extensas, colapso dos alvéolos e pontuações de Ashcroft mais altas que quantificam a fibrose. Genes envolvidos na produção de colágeno e outras vias relacionadas à fibrose estavam mais ativos nesses camundongos knockout, confirmando que a perda de Kdm7a torna os pulmões mais vulneráveis à formação de cicatrizes duradouras e prejudiciais.
Como KDM7A afasta os macrófagos de um destino promotor de cicatrizes
Usando sequenciamento de RNA de célula única, os autores ampliaram o foco para células individuais dos pulmões de camundongos lesionados. Descobriram que, na ausência de Kdm7a, um subconjunto particular de macrófagos no tecido de sustentação do pulmão se expandiu dramaticamente e adquiriu uma forte assinatura Fib-Mac, expressando genes como Arg1, Spp1 e Trem2. Experimentos adicionais em macrófagos cultivados mostraram que a remoção de Kdm7a aumentou os genes marcadores de Fib-Mac e reprogramou o metabolismo celular em direção a vias que sustentam a produção de colágeno e a ativação prolongada. Em outras palavras, KDM7A normalmente restringe tanto os programas genéticos quanto os metabólicos que impulsionam os macrófagos a um estado promotor de fibrose.
Aprofundando-se, os pesquisadores identificaram um parceiro-chave nesse sistema de freio: uma proteína sensora chamada TLR8, que detecta fragmentos de RNA dentro das células imunes. Eles descobriram que KDM7A ajuda a manter o gene Tlr8 ativado removendo uma marca química repressora (H3K27me2) de uma região potenciadora próxima a Tlr8. Quando Kdm7a foi desativado, essa marca se acumulou, os níveis de Tlr8 caíram e as características Fib-Mac se intensificaram. Reduzir diretamente Tlr8 em macrófagos também os empurrou em direção a uma identidade fibrótica, enquanto ativar ou superexpressar TLR8 os trouxe de volta, mesmo quando Kdm7a estava ausente. Isso coloca a via KDM7A–TLR8 no centro de um circuito molecular que protege os pulmões contra cicatrização excessiva.
Dos pulmões envelhecidos à doença humana
Para conectar essas descobertas às pessoas, a equipe examinou tecido pulmonar de pacientes com doença pulmonar fibrótica. Em comparação com tecido controle não doente, pulmões fibróticos continham muito mais macrófagos portando marcadores Fib-Mac, mas essas mesmas células mostraram níveis marcadamente reduzidos de KDM7A e TLR8. Reanálises de conjuntos de dados de célula única existentes de pacientes com fibrose pulmonar idiopática confirmaram esse padrão: à medida que as assinaturas Fib-Mac aumentavam, a expressão de KDM7A caía. Os pesquisadores também exploraram um grande atlas de camundongos e descobriram que a expressão de Kdm7a e Tlr8 em macrófagos declinava com a idade em machos, refletindo o maior risco de fibrose pulmonar em homens mais velhos. Isso sugere que o enfraquecimento relacionado à idade e ao sexo do freio KDM7A–TLR8 pode ajudar a explicar quem é mais vulnerável à formação severa de cicatrizes pulmonares.
O que isso significa para tratamentos futuros
Em termos simples, este trabalho mostra que nosso sistema imunológico carrega um mecanismo interno de segurança que impede que células reparadoras úteis se tornem excessivas e se transformem em motoristas de cicatrizes permanentes. KDM7A, atuando por meio de TLR8, impede que os macrófagos se prendam a um modo profibrótico e, assim, ajuda a manter o tecido pulmonar flexível e funcional após uma lesão. Quando esse sistema falha — por perda genética, envelhecimento ou outros fatores — os macrófagos têm maior probabilidade de se tornar “amplificadores de cicatrizes”, agravando a fibrose. Ao revelar esse freio epigenético, o estudo aponta para novas estratégias terapêuticas: medicamentos que aumentem a atividade de KDM7A, imitem seus efeitos ou estimulem cuidadosamente TLR8 podem, um dia, complementar as terapias antifibróticas existentes e oferecer melhor proteção contra a cicatrização progressiva e limitante da vida dos pulmões.
Citação: Funagura, N., Koga, T., Etoh, K. et al. Histone demethylase KDM7A negatively regulates fibrotic macrophage polarization and lung fibrosis progression. Commun Biol 9, 309 (2026). https://doi.org/10.1038/s42003-026-09610-1
Palavras-chave: fibrose pulmonar, macrófagos, epigenética, KDM7A, TLR8