Clear Sky Science · pt
haCCA: integração multimódulo de transcriptomas e metabolomas espaciais baseados em pontos
Por que mapear moléculas no lugar importa
Nossos corpos são formados por incontáveis pequenos bairros de células, cada um com sua própria combinação de genes ativos e substâncias químicas. Até recentemente, os cientistas precisavam estudar essas moléculas após triturar o tecido em uma pasta uniforme, perdendo totalmente a noção de “onde” as coisas estavam. Este artigo apresenta um novo método computacional, chamado haCCA, que costura duas técnicas de imagem poderosas para que os pesquisadores possam ver, in situ, como genes e pequenas moléculas se organizam em tecidos e tumores reais. Esse tipo de mapa pode revelar padrões ocultos da doença e sugerir tratamentos mais precisos.
DuAS vistas diferentes do mesmo tecido
O estudo concentra-se na combinação de dados de dois métodos espaciais cada vez mais usados em biologia. A transcriptômica espacial registra quais genes estão ligados em milhares de pequenos pontos em uma fatia de tecido. A imagem por espectrometria de massa MALDI registra as quantidades de muitas pequenas moléculas, como metabólitos e lipídios, em grades de pontos igualmente densas. O problema é que esses dois instrumentos não medem as mesmas posições exatas nem o mesmo conjunto de características, então seus dados são como dois mapas desalinhados com legendas diferentes. Abordagens existentes tentam principalmente corresponder as formas das seções de tecido com base apenas em suas coordenadas, o que pode ser impreciso e não oferece uma maneira de verificar quão bem o alinhamento realmente funcionou.
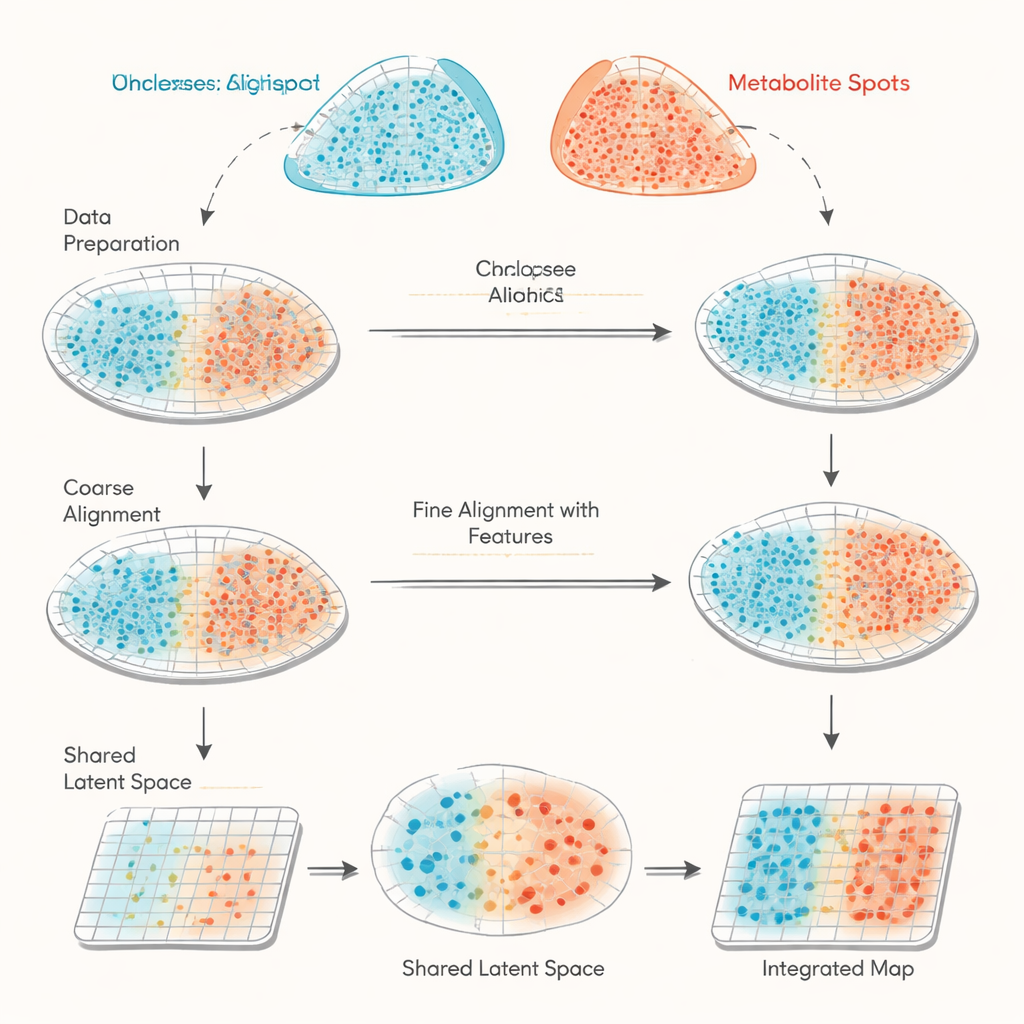
Uma forma mais inteligente de alinhar mapas moleculares
haCCA (sigla para análise de correlação canônica guiada por âncoras hierárquicas) enfrenta esse desafio combinando geometria com biologia. Primeiro, executa um “alinhamento morfológico” em duas etapas das grades de pontos das duas tecnologias. Especialistas humanos escolhem alguns marcos correspondentes nas imagens do tecido para corrigir aproximadamente deslocamentos e rotações, e então uma etapa automatizada ajusta finamente outliers próximos a bordas rasgadas ou peças ausentes. Em seguida, o método procura pares de “âncoras” de pontos que estejam próximos no espaço e localizados em regiões localmente uniformes, tornando provável que representem a mesma área do tecido. A partir desses pontos âncora, haCCA calcula quais genes e metabólitos tendem a variar juntos e os destila em uma representação compartilhada de baixa dimensionalidade que captura seus padrões conjuntos mais fortes.
Transformando correlações em uma imagem unificada do tecido
Com as coordenadas espaciais e a representação molecular compartilhada em mãos, haCCA resolve um problema de otimização para decidir quão provável é que cada ponto de gene deva ser pareado com cada ponto de metabólito. Esta etapa é projetada para manter pontos próximos no espaço, mas também semelhantes em seu perfil combinado gene–metabólito. O resultado final é um “plano de transporte” que liga cada ponto de um conjunto de dados ao seu melhor parceiro no outro, produzindo um mapa multimodal integrado. Em dados de teste cuidadosamente construídos — onde as relações verdadeiras são conhecidas — os autores mostram que cada estágio do fluxo de trabalho (alinhamento grosseiro, alinhamento refinado e correspondência sensível a características) melhora progressivamente três medidas independentes de precisão. Em comparação com outras ferramentas que dependem principalmente da geometria, haCCA alcança consistentemente maior alinhamento e transferência mais fiel dos rótulos de região.
Revelando biologia oculta em cânceres de cérebro e fígado
Os autores aplicam então haCCA a tecidos reais de cérebro de camundongo e tumores hepáticos. Para o cérebro, eles integram dados comerciais de transcriptômica espacial com imagens de metabólitos das mesmas seções ou de seções vizinhas. O método preserva territórios metabólicos conhecidos e reconstrói sobreposições esperadas, como a colocalização da dopamina com o gene que codifica sua enzima chave. Ao agrupar conjuntamente genes e metabólitos, eles descobrem que os dados combinados distinguem sub-regiões de tecido mais nuanceadas do que cada modalidade isoladamente. Em um modelo pré-clínico de colangiocarcinoma intra-hepático, um tipo de câncer de fígado, usam haCCA para comparar tumores que podem ou não formar armadilhas extracelulares de neutrófilos — estruturas em forma de rede liberadas por células imunes. Os mapas integrados revelam que, quando essas armadilhas estão presentes, um gene chamado Scd1 e seus ácidos graxos associados estão enriquecidos em regiões malignas, apontando para uma mudança em direção a um metabolismo de lipídios alterado no tumor.
O que isso significa para pesquisas futuras
Em termos cotidianos, haCCA é como alinhar fotos aéreas tiradas com câmeras diferentes — uma sensível a contornos de prédios, a outra a assinaturas térmicas — para obter uma imagem mais nítida do que está acontecendo em cada quarteirão. Ao fundir com precisão onde os genes estão ativos com onde metabólitos-chave se acumulam, esse fluxo de trabalho ajuda os cientistas a perfilar simultaneamente os dois lados do comportamento celular: as instruções e a química resultante. A abordagem melhora métodos de alinhamento anteriores, está empacotada em uma ferramenta Python acessível e pode ser estendida a outras tecnologias espaciais. À medida que mapas integrados assim se tornarem mais rotineiros, eles poderão aprofundar nossa compreensão de como tumores e outros tecidos organizam seu metabolismo, respondem a tratamentos e evoluem ao longo do tempo.
Citação: Xu, J., Shen, XT., Zhang, C. et al. haCCA: multi-module Integration of spot-based spatial transcriptomes and metabolomes. Commun Biol 9, 248 (2026). https://doi.org/10.1038/s42003-026-09526-w
Palavras-chave: multiômica espacial, transcriptômica, metabolômica, metabolismo tumoral, integração de dados