Clear Sky Science · pt
Determinação ab initio da estabilidade de fases em sólidos com desordem dinâmica: desordem rotacional C2 em Li2C2
Por que esse sólido mutável importa
Muitas tecnologias modernas dependem de sólidos que podem modificar discretamente sua estrutura interna quando aquecidos ou comprimidos. Essas mudanças, chamadas transições de fase, são centrais em ideias como refrigeração sólida e baterias mais seguras. Este estudo examina um composto simples, o carbeto de lítio (Li2C2), que passa de uma forma ordenada e bem definida para uma forma mais agitada e dinamicamente desordenada conforme a temperatura aumenta. Ao acompanhar essa transformação átomo a átomo em simulações computacionais, os autores mostram como a “irritabilidade” interna de pequenas unidades moleculares pode deslocar o equilíbrio entre duas estruturas cristalinas.
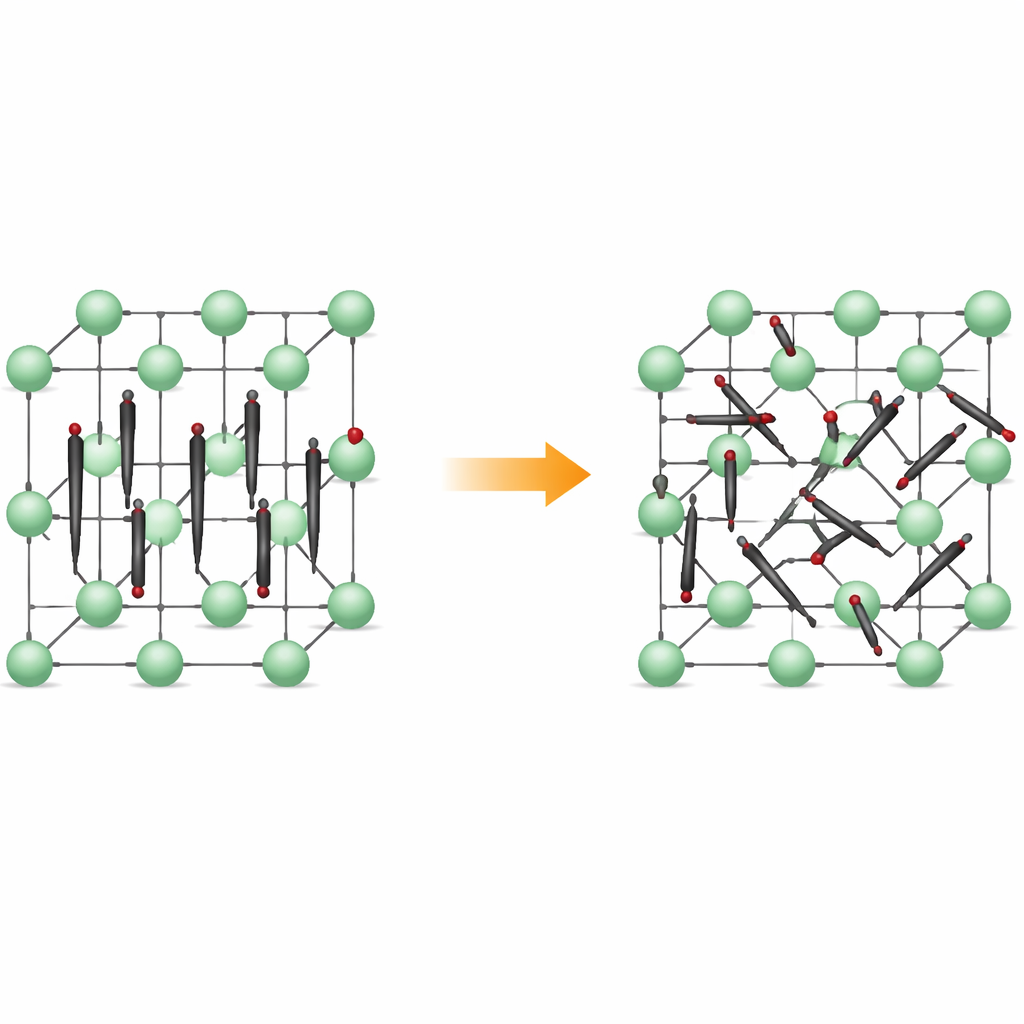
De fileiras organizadas ao movimento inquieto
Em baixas temperaturas, o Li2C2 forma um cristal ortorrômbico: seus átomos de carbono agrupam-se em pequenos dímeros C2 que apontam quase todos na mesma direção, como palitos de fósforo alinhados. Íons de lítio ficam intercalados, criando uma malha tridimensional regular. Quando o material é aquecido, ele se transforma numa forma cúbica, onde as posições dos centros dos dímeros permanecem ordenadas numa rede, mas os próprios dímeros deixam de manter uma direção fixa. Em vez disso, eles rotacionam entre várias orientações preferenciais, passando tempo em vales de energia rasos que correspondem a alinhamentos específicos. O material continua sólido, mas sua estrutura interna torna-se dinamicamente desordenada.
Acompanhando a mudança ao longo de um caminho suave
Para entender qual fase é mais estável a uma dada temperatura, é preciso comparar suas energias livres, que combinam energia e entropia (uma medida de desordem). Métodos padrão baseados em pequenas vibrações ao redor de posições fixas enfrentam dificuldades quando átomos se deslocam ou rotacionam significativamente. Aqui, os autores usam uma técnica chamada integração termodinâmica tensão-deformação, apoiada em dinâmica molecular de primeira-princípios. Eles constroem um caminho de deformação suave que transforma continuamente a célula de simulação da estrutura ortorrômbica de baixa temperatura para a cúbica de alta temperatura. Ao longo desse caminho, realizam longas simulações a temperaturas fixas e medem como a tensão interna responde à deformação imposta. Integrar essa resposta de tensão fornece a diferença de energia livre entre as duas fases.
Ver a entropia através do movimento atômico
Os cálculos revelam que por volta de 600 K a fase ortorrômbica de baixa temperatura ainda é ligeiramente favorecida, enquanto a 650 K a fase cúbica vence por alguns milésimos de elétron-volt por fórmula unitária. Interpolando esses resultados obtém-se uma temperatura de transição de aproximadamente 611 K. Isso é menor que estimativas experimentais, mas ainda em acordo razoável, dado as pequenas diferenças de energia livre envolvidas. A energia interna da fase cúbica é na verdade maior; o que a estabiliza é um grande ganho de entropia, rastreado diretamente até a desordem rotacional dos dímeros C2. Ao analisar como a orientação de cada dímero perde a memória de sua direção inicial ao longo do tempo, os autores mostram que os dímeros se reorientam em escalas de tempo sub-picosegundo, borrando a linha entre as categorias usuais de entropia “vibracional” e “configuracional”.
Além das imagens simples de desordem sólida
O trabalho também destaca que atalhos comuns — como tratar a entropia como a soma simples de vibrações em torno de configurações fixas mais a contagem separada de orientações estáticas — falham para materiais como o Li2C2. Como as rotações dos dímeros são rápidas e fortemente acopladas às vibrações ordinárias, o sistema não pode ser claramente dividido em partes “vibrantes” e “rearranjantes”. O método de integração tensão-deformação contorna essa dificuldade: ele extrai a energia livre total diretamente da dinâmica microscópica, sem precisar supor como a entropia deve ser repartida.
O que o estudo nos ensina
Em termos cotidianos, o estudo mostra como um sólido pode permanecer rígido enquanto seus blocos de construção internos se tornam progressivamente livres para girar e se mover, e como essa liberdade interna pode tornar uma estrutura mais desordenada termodinamicamente preferível. Para o Li2C2, a fase cúbica de alta temperatura é estabilizada não porque seja energeticamente mais barata, mas porque oferece muito mais modos para os dímeros C2 se orientarem e se movimentarem. Ao demonstrar que a integração termodinâmica tensão-deformação pode captar esse equilíbrio sutil entre ordem, energia e entropia, o trabalho abre caminho para prever transições semelhantes em outros sólidos com desordem dinâmica que podem sustentar futuros dispositivos de refrigeração, baterias e materiais inteligentes.
Citação: Klarbring, J., Filippov, S., Häussermann, U. et al. Ab initio determination of phase stabilities of dynamically disordered solids: rotational C2 disorder in Li2C2. Sci Rep 16, 8965 (2026). https://doi.org/10.1038/s41598-026-43795-z
Palavras-chave: transição de fase no estado sólido, desordem dinâmica, dynamics molecular, carbetos de lítio, integração termodinâmica