Clear Sky Science · pt
Projeto orientado por dados de bloqueadores LNA para remoção eficiente de contaminantes em bibliotecas de Ribo-Seq
Por que a limpeza dos dados de sequenciamento é importante
A biologia moderna costuma depender da leitura de milhões de pequenos fragmentos de RNA para entender como as células produzem proteínas. Mas essas medidas poderosas, especialmente um método chamado perfilagem de ribossomos (Ribo‑Seq), podem ficar poluídas por trechos de RNA irrelevantes que desperdiçam capacidade de sequenciamento e dinheiro. Este estudo descreve uma maneira simples, orientada por dados, de projetar “bloqueadores” moleculares especializados que removem seletivamente esses fragmentos indesejados, quase dobrando a informação útil que os pesquisadores obtêm do mesmo experimento.
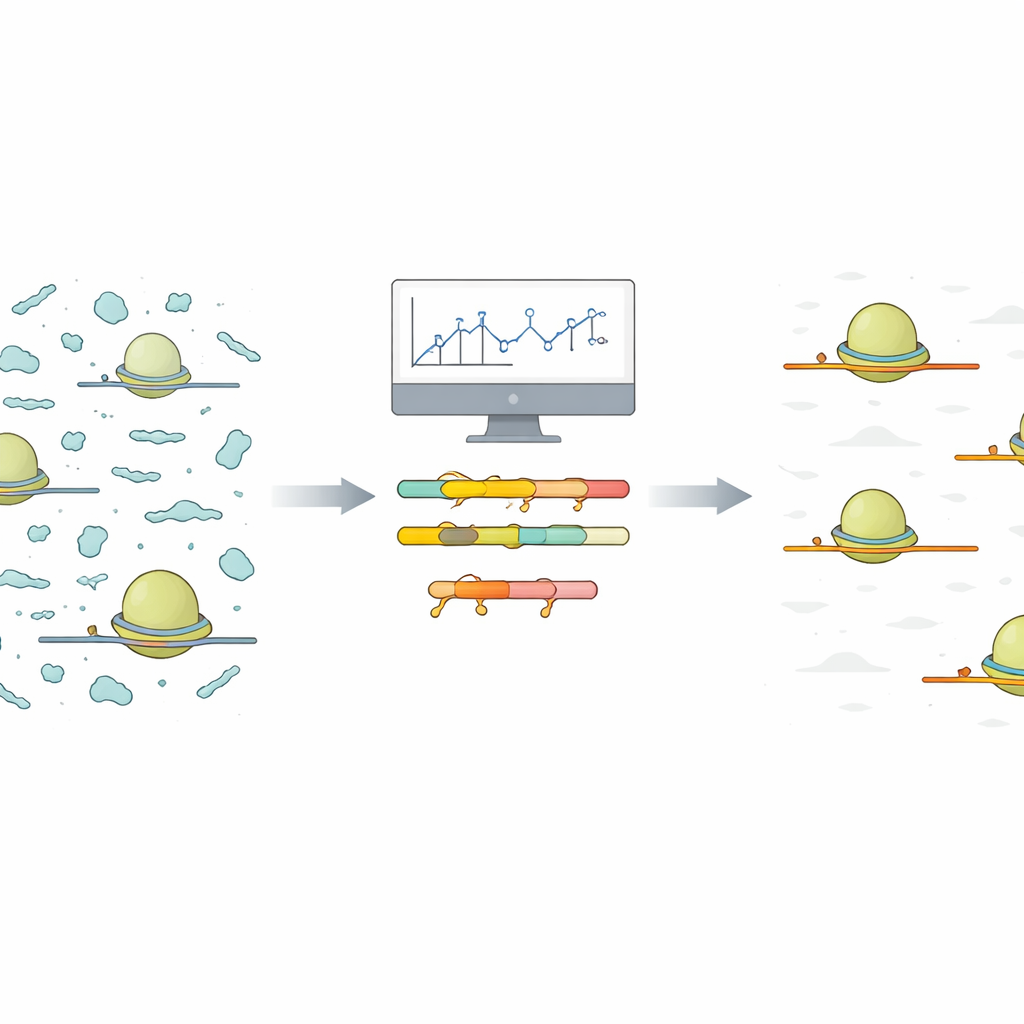
O problema dos instantâneos ruidosos de ribossomos
Ribo‑Seq captura um instantâneo momento a momento de quais mensagens em uma célula estão sendo ativamente traduzidas em proteínas. Para isso, os cientistas isolam ribossomos junto com os pequenos trechos de RNA mensageiro (mRNA) que eles protegem. Todo o resto é degradado, e os trechos protegidos são sequenciados e mapeados de volta ao genoma. Na prática, entretanto, muitos outros pequenos pedaços de RNA não codificante passam por esse processo. Como esses fragmentos contaminantes são abundantes e altamente variáveis, eles consomem uma grande fração das leituras de sequenciamento, deixando menos leituras para os sinais verdadeiros codificadores de proteína que interessam aos pesquisadores.
Por que os truques de limpeza existentes ficam aquém
As estratégias padrão tentam remover RNAs ribossomais abundantes e outros RNAs não codificantes com sondas de captura pré-projetadas ou enzimas. Esses métodos funcionam bem quando os RNAs-alvo estão intactos e previsíveis, mas o Ribo‑Seq propositalmente fragmenta o RNA em muitos pedaços de tamanhos diferentes. Essa fragmentação embaralha os locais-alvo para conjuntos de sondas fixas, tornando a depleção muito menos eficiente. Além disso, a mistura exata de contaminantes depende da espécie estudada, das condições de crescimento e até de qual nuclease é utilizada. Fluxos de trabalho de limpeza existentes também tendem a envolver múltiplas etapas de incubação e purificação, que consomem tempo e podem causar perda de amostra ou viés.
Bloqueadores personalizados projetados a partir de dados reais
Os autores propõem uma abordagem simplificada que começa com uma corrida de sequenciamento piloto pequena e de baixo custo nas mesmas condições planejadas para o experimento completo. Eles fornecem um script em R que recebe as leituras alinhadas desse ensaio piloto e agrupa automaticamente fragmentos contaminantes semelhantes com base na sequência. Para cada grupo, o script informa a menor sequência comum que aparece nos fragmentos. Esses trechos curtos e compartilhados são sítios-alvo ideais para moléculas especializadas chamadas oligonucleotídeos de ácido nucleico travado (LNA). LNAs são fitas curtas com uma modificação química que as faz se ligar muito fortemente ao RNA correspondente. O script também gera mapas de calor e gráficos resumo intuitivos, ajudando os usuários a ver quais contaminantes dominam e quantos alvos LNA seriam necessários para uma limpeza substancial.
Uma limpeza em um passo durante a amplificação
Em vez de remover fisicamente os contaminantes da amostra, o método usa oligonucleotídeos LNA como bloqueadores durante a etapa de amplificação do DNA que constrói a biblioteca de sequenciamento. Os autores testaram a adição desses bloqueadores tanto durante a etapa inicial de transcrição reversa quanto durante a amplificação por PCR posterior. Eles descobriram que a adição de LNAs durante a amplificação foi mais eficiente e exigiu concentrações menores, reduzindo um contaminante teste em mais de mil vezes e funcionando independentemente da orientação de cadeia. Dicas práticas de projeto incluem alternar blocos construtores de DNA padrão e LNA, usar comprimento mínimo de 14 unidades para a planta Arabidopsis, e modificar a extremidade terminal para que o próprio bloqueador não possa ser acidentalmente estendido.
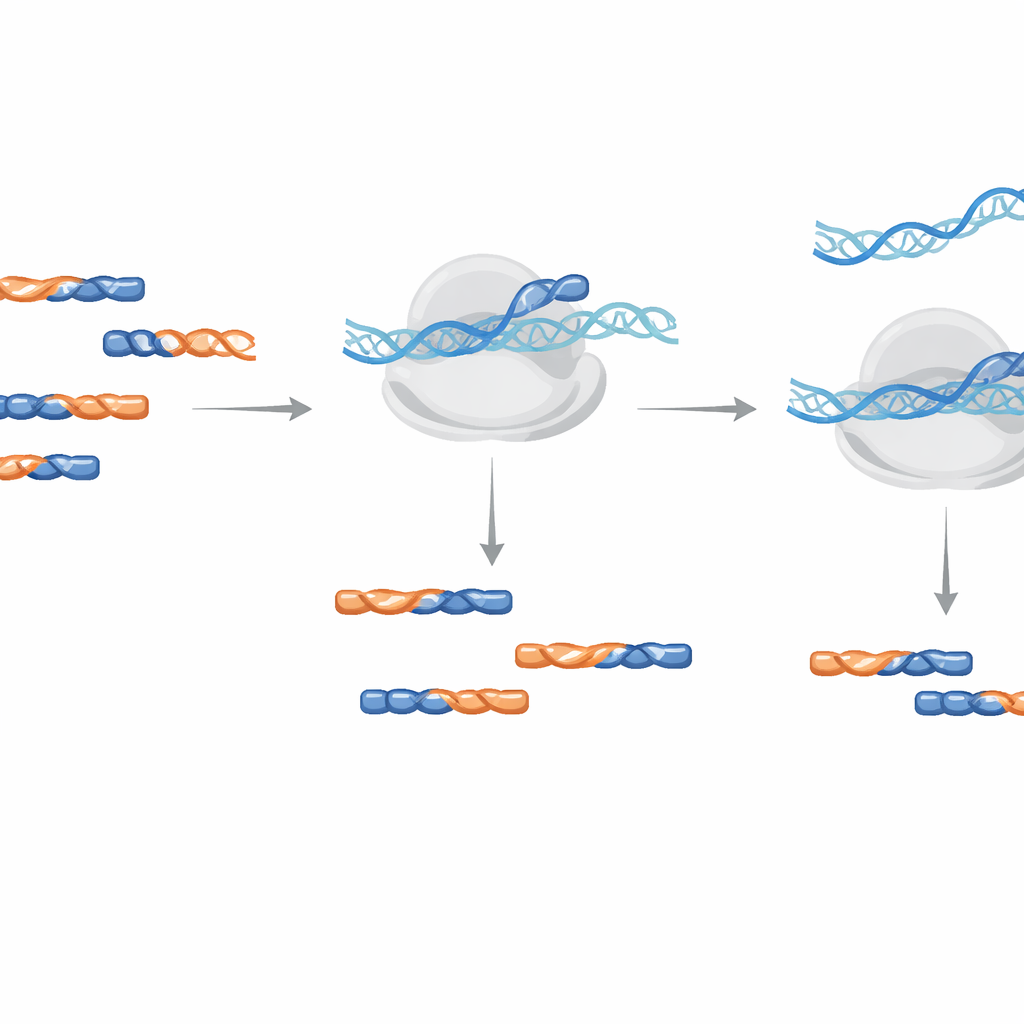
Mais leituras úteis sem distorcer o sinal
Para demonstrar o desempenho no mundo real, a equipe projetou cinco bloqueadores LNA direcionados aos grupos de contaminantes mais comuns observados em condições típicas de crescimento em plantas de Arabidopsis. Quando adicionaram essa mistura durante a amplificação da biblioteca, a proporção de contaminantes identificados caiu mais de 30%, e o número de leituras úteis codificadoras de proteína quase dobrou. Crucialmente, quando compararam as contagens de leitura ao nível de gene entre bibliotecas com e sem tratamento com LNA, os valores concordaram quase perfeitamente, indicando que os bloqueadores removeram fragmentos de sucata sem distorcer o sinal biológico proveniente dos verdadeiros footprints de mRNA.
O que isso significa para experimentos futuros
Este trabalho mostra que um experimento piloto curto, combinado com um script de análise fácil de usar e um pequeno conjunto de bloqueadores LNA sob medida, pode transformar bibliotecas Ribo‑Seq bagunçadas em conjuntos de dados muito mais limpos e informativos em um único passo de pipetagem. Os pesquisadores ganham mais leituras significativas por execução, economizando custos e simplificando o desenho experimental, enquanto preservam medições precisas de como os genes são traduzidos. Os autores também fornecem perfis de contaminantes prontos e projetos de bloqueadores para condições vegetais comuns, e sugerem que recursos semelhantes poderiam ser construídos para muitos organismos, tornando a perfilagem de ribossomos de alta qualidade mais acessível à comunidade científica.
Citação: Ricciardi, D.A., Peter, F.E. & Böhmer, M. Data-driven design of LNA-blockers for efficient contaminant removal in Ribo-Seq libraries. Sci Rep 16, 8565 (2026). https://doi.org/10.1038/s41598-026-43117-3
Palavras-chave: perfilagem de ribossomos, contaminantes de RNA, ácidos nucleicos travados (LNA), limpeza de bibliotecas de sequenciamento, regulação da tradução