Clear Sky Science · pt
Modelo híbrido de deep learning ajustado para diagnóstico de câncer de mama usando dados genéticos
Por que isso importa para pacientes e famílias
O câncer de mama é atualmente o tipo de câncer mais diagnosticado em mulheres no mundo, e detectá-lo precocemente pode ser a diferença entre a vida e a morte. Os médicos têm cada vez mais acesso às informações genéticas de uma pessoa, mas transformar dezenas de milhares de medições gênicas em respostas claras é extraordinariamente difícil. Este artigo descreve um novo modelo computacional que lê esses padrões genéticos complexos para detectar câncer de mama e prever desfechos com precisão notável, potencialmente fornecendo aos clínicos um assistente poderoso para decisões mais precoces e confiáveis.
Dos genes aos sinais de alerta
Cada tumor de mama carrega uma impressão digital molecular codificada na atividade de milhares de genes. Os autores propuseram construir um sistema capaz de ler essa impressão digital diretamente, em vez de depender apenas de imagens ou de um punhado de genes bem conhecidos, como BRCA1 e BRCA2. Trabalharam com dois dos maiores recursos públicos em genômica do câncer: a coorte de câncer de mama do TCGA, que inclui atividade gênica para 17.814 genes em 590 amostras, e o estudo METABRIC, que contém informações genômicas e clínicas de mais de 1.400 pacientes. O objetivo deles foi ambicioso: projetar um método que consiga lidar com esse fluxo de informação, encontrar os sinais mais reveladores e ainda funcionar de forma confiável em grupos de pacientes completamente independentes.
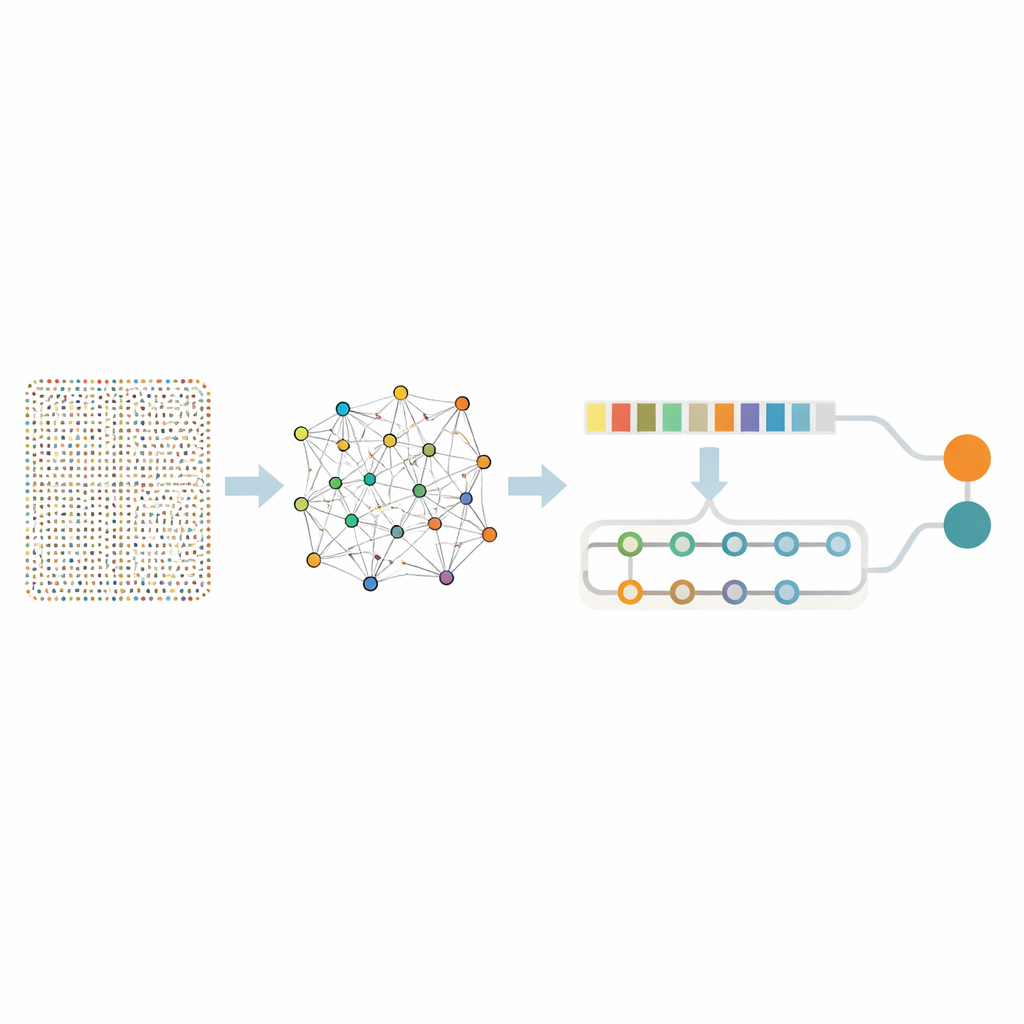
Reduzindo milhares de genes a um conjunto útil
Analisar quase dezoito mil genes de uma vez é excessivo mesmo para algoritmos avançados, e isso corre o risco de captar ruído sem significado. Os pesquisadores, portanto, usaram um “peneiramento” em duas etapas para isolar um conjunto menor de genes realmente informativos. Primeiro, aplicaram uma técnica chamada Random Forest, que basicamente pergunta a muitas árvores de decisão quais genes são mais importantes para distinguir tecido canceroso de amostras saudáveis. Essa etapa reduziu a lista para 436 genes promissores. Em seguida, examinaram como esses genes se comportam em conjunto usando mineração de regras de associação, um método que identifica grupos de genes que tendem a estar ativos ao mesmo tempo em tumores. Essa camada extra de análise identificou pares e redes de genes ligados a processos-chave do câncer, como divisão celular acelerada, reparo de dano ao DNA e alterações no tecido ao redor do tumor. Após esse refinamento, permaneceram 332 genes — ainda ricos em significado biológico, mas muito mais manejáveis para análises mais profundas.
Uma rede neural em duas partes que aprende padrões e contexto
Com esse conjunto focado de genes em mãos, a equipe construiu um modelo híbrido de deep learning que combina dois tipos de redes neurais. Uma parte, conhecida como rede convolucional, varre a lista de genes para captar padrões locais — aglomerados de genes que tendem a subir ou cair juntos. A segunda parte, uma rede de memória bidirecional, olha para a mesma informação mantendo o rastreamento de relações de longo alcance, capturando como genes distantes influenciam uns aos outros ao longo de todo o perfil. Antes do treinamento, os autores equilibraram os dados para que amostras com e sem câncer estivessem representadas de forma justa e adicionaram pequenas quantidades de ruído artificial, ensinando o modelo a não ser enganado por flutuações aleatórias.
Como o sistema se sai em testes do mundo real
Quando treinada e testada nos dados do TCGA, a rede híbrida distinguiu corretamente amostras tumorais de normais com cerca de 97% de acurácia e uma capacidade quase perfeita de separar os dois grupos. Importante, superou configurações mais simples de deep learning e ferramentas padrão de machine learning, como regressão logística e máquinas de vetores de suporte, mesmo quando esses métodos concorrentes receberam os mesmos genes cuidadosamente selecionados. O teste mais exigente, porém, foi ver se o modelo se manteria em um conjunto de dados totalmente diferente. Aplicado ao METABRIC, coletado em outros hospitais usando métodos laboratoriais distintos, o sistema manteve alto desempenho: em sua melhor execução alcançou 99,3% de acurácia e identificou corretamente todos os pacientes que posteriormente morreram de câncer de mama, uma propriedade crucial se a ferramenta for usada para sinalizar casos de alto risco.
O que isso pode significar para cuidados futuros
Para um não especialista, a conclusão é que este estudo entrega um filtro e leitor inteligente para dados genéticos que pode detectar câncer de mama e risco relacionado com notável consistência em grandes grupos de pacientes. Ao combinar uma estratégia cuidadosa de seleção de genes com uma rede neural de dois ramos, os autores mostram que computadores podem extrair sinais clinicamente significativos de enormes conjuntos de dados genéticos, não apenas em um estudo, mas em coortes independentes. Embora sejam necessários mais trabalhos para testar a abordagem em populações diversas e explicar suas decisões em detalhes, o método aponta para um futuro no qual uma simples amostra de sangue ou tecido poderia alimentar tais modelos e ajudar médicos a detectar tumores mais cedo e a ajustar tratamentos com mais precisão.
Citação: Hesham, F., Abbassy, M.M. & Abdalla, M. Hybrid tuned deep learning model for breast cancer diagnosis using genetic data. Sci Rep 16, 9664 (2026). https://doi.org/10.1038/s41598-026-41643-8
Palavras-chave: genômica do câncer de mama, diagnóstico por deep learning, biomarcadores de expressão gênica, detecção precoce do câncer, suporte à decisão clínica