Clear Sky Science · pt
DPAS: pontuação de anomalia de peptídeos associados a doenças para identificar peptídeos patogênicos via aprendizado one-class
Por que pequenas partes de proteínas importam para nossa saúde
Peptídeos — curtos trechos de proteínas — tornaram-se protagonistas crescentes na medicina contemporânea. Eles podem agir como mensageiros precisos no organismo e são cada vez mais usados como fármacos e marcadores de doença. Ainda assim, descobrir quais peptídeos estão realmente ligados a doenças geralmente depende de ter exemplos claros tanto de peptídeos “doentes” quanto de peptídeos “não-doentes”, algo que a biologia raramente fornece. Este estudo introduz uma nova forma de identificar peptídeos potencialmente nocivos usando apenas aqueles que já sabemos estar envolvidos em doença, oferecendo um caminho mais rápido e menos tendencioso para descobrir futuros diagnósticos e tratamentos.
O desafio de encontrar o grupo “não-doente”
Modelos computacionais tradicionais aprendem comparando dois lados: exemplos positivos conhecidos por estarem relacionados à doença e exemplos negativos que se supõe serem inofensivos. Na pesquisa com peptídeos, esse segundo grupo é problemático. Muitos peptídeos simplesmente não foram testados, de modo que rotulá‑los como “não-doentes” pode ser enganoso e introduzir viés. Estudos anteriores sobre peptídeos anticâncer ou anti-inflamatórios alcançaram precisão impressionante, mas frequentemente dependiam de conjuntos de dados negativos construídos manualmente ou supostos. Como resultado, seus modelos podem ter dificuldade com sinais raros ou novos tipos de peptídeos patológicos que não se assemelham aos dados de treinamento.
Aprendendo com o que sabemos, em vez do que supomos
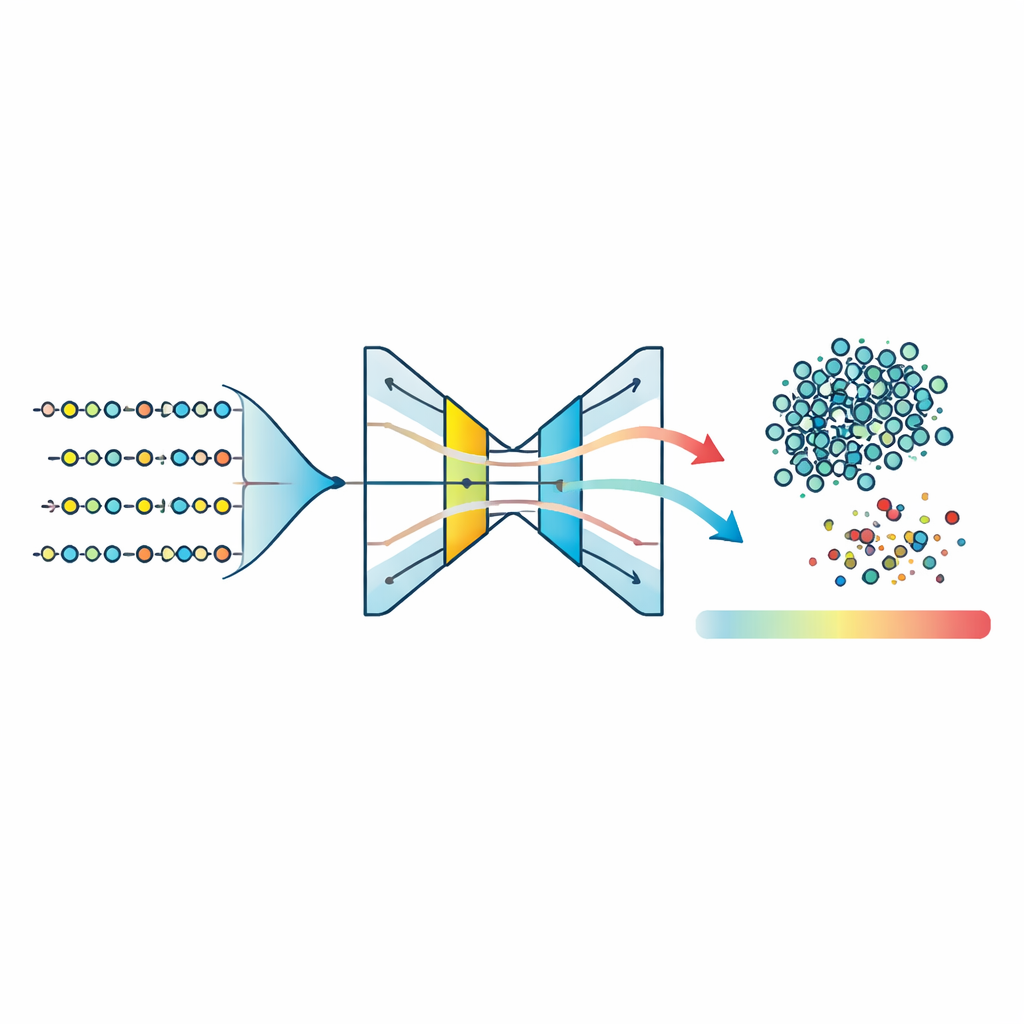
Os autores seguem um caminho diferente: em vez de forçar um problema de dois lados, tratam os peptídeos associados à doença como um grupo coerente e perguntam: “Como esse grupo se parece em detalhe?” Eles coletam mais de 760.000 peptídeos humanos mutados a partir de um banco de dados especializado em câncer e descrevem cada peptídeo usando um conjunto rico de características. Isso inclui a frequência de cada aminoácido, a disposição de pares de aminoácidos, traços físico‑químicos básicos como volume e afinidade por água, e padrões curtos recorrentes de sequência conhecidos como motivos. Uma técnica chamada análise de componentes principais (PCA) então comprime essa descrição de alta dimensão para uma forma mais manejável, preservando as principais fontes de variação.
Identificando peptídeos incomuns com modelos one-class
Com esse espaço de características comprimido em mãos, a equipe treina três modelos “one-class” — algoritmos projetados para aprender a forma de um único grupo e sinalizar tudo o que não se encaixa. Eles testam One-Class Support Vector Machines, Isolation Forests e um tipo de rede neural chamada autoencoder. O autoencoder aprende a comprimir as características de cada peptídeo para uma representação interna estreita e depois reconstruí‑las; peptídeos que pertencem ao padrão de doença aprendido são reconstruídos com precisão, enquanto os incomuns apresentam erro de reconstrução mais alto. Comparando scores de anomalia normalizados entre todos os métodos, observa‑se que o autoencoder produz o aglomerado mais compacto de peptídeos típicos e a separação mais clara entre inliers e outliers. Ao definir um limiar no erro de reconstrução em torno do percentil 95, o modelo classifica a maioria dos peptídeos como provavelmente associados à doença enquanto sinaliza de forma consistente uma pequena fração como atípica.
Convertendo scores complexos em um número único e significativo
Para tornar os resultados mais fáceis de interpretar biologicamente, os autores introduzem a Pontuação de Anomalia de Peptídeos de Doença (DPAS). Essa pontuação mistura dois ingredientes: quão incomum um peptídeo parece para o autoencoder (seu erro de reconstrução normalizado) e quão fortemente suas características contribuem para as previsões, medido por um método de explicação popular chamado SHAP. Na prática, motivos e traços físico‑químicos específicos emergem como especialmente informativos. O DPAS combina esses sinais de modo que peptídeos que são estruturalmente estranhos e apoiados por características biologicamente relevantes recebam classificações mais altas. Os peptídeos com maiores pontuações são então examinados com uma ferramenta de busca de motivos que os associa a assinaturas funcionais conhecidas, como locais de fosforilação, regiões de ligação a metais e outros padrões regulatórios comumente envolvidos em sinalização e controle enzimático.
O que isso significa para diagnósticos e medicamentos futuros
Em termos práticos, este trabalho oferece um filtro mais inteligente para encontrar peptídeos suspeitos sem fingir que sabemos quais são definitivamente inofensivos. Ao aprender apenas com exemplos confirmados relacionados à doença e depois ranquear novos candidatos com o DPAS, os pesquisadores podem priorizar uma lista curta e biologicamente plausível de peptídeos para testes laboratoriais. Muitos dos candidatos melhor classificados contêm motivos funcionais bem conhecidos, reforçando a ideia de que podem desempenhar papéis em processos patológicos. Embora o método ainda dependa de suposições e careça de peptídeos “seguros” validados experimentalmente para uma validação completa, fornece uma base mais realista e transparente para a descoberta de biomarcadores peptídicos e pode ser adaptado a outros tipos de dados biológicos onde exemplos negativos confiáveis são escassos.
Citação: Khalid, Z., Khalid, R. & Sezerman, O.U. DPAS: disease-associated peptide anomaly score for identifying pathogenic peptides via one-class learning. Sci Rep 16, 9170 (2026). https://doi.org/10.1038/s41598-026-40099-0
Palavras-chave: peptídeos associados a doenças, detecção de anomalias, autoencoder, descoberta de biomarcadores, aprendizado one-class