Clear Sky Science · pt
Aprendizagem de máquina interpretável racionaliza a inibição da anidrase carbônica por meio de predição conformal e contrafactual
Por que medicamentos contra o câncer mais inteligentes importam
Medicamentos contra o câncer frequentemente atuam como ferramentas pouco precisas: embora ataquem células tumorais, também podem atingir tecidos saudáveis e causar efeitos colaterais sérios. Uma forma promissora de afiar essa precisão é bloquear versões específicas de uma enzima chamada anidrase carbônica, que ajuda os tumores a sobreviver em ambientes com pouco oxigênio. Porém, várias isoformas dessa enzima são quase idênticas, tornando difícil projetar fármacos que atinjam as variantes “ruins” nos tumores sem afetar a isoforma “boa” presente por todo o corpo. Este estudo mostra como aprendizagem de máquina interpretável pode ajudar pesquisadores a navegar esse desafio e projetar candidatos a fármacos mais seletivos e seguros.
O problema de atingir o alvo errado
A anidrase carbônica humana (hCA) existe em muitas formas, ou isoformas. Duas delas, IX e XII, estão ligadas à sobrevivência de células cancerosas em tumores com falta de oxigênio, de modo que bloqueá‑las pode retardar a doença e melhorar o tratamento. Mas a isoforma II é generalizada em tecidos saudáveis e tem um sítio ativo muito parecido com os de IX e XII. Fármacos que se ligam às três podem desencadear problemas indesejados, como acidose metabólica e distúrbios visuais. Métodos tradicionais de laboratório e computacionais têm dificuldade porque as enzimas são moléculas grandes e complexas, e o número de compostos com características de fármaco possíveis é astronomicamente alto. Testá‑los exaustivamente, seja em laboratório ou por computador, simplesmente não é viável.
Construindo uma base de dados limpa e confiável
Os autores abordaram isso reunindo primeiro um banco de dados cuidadosamente limpo com milhares de moléculas testadas contra hCA II, IX e XII a partir do repositório ChEMBL. Eles padronizaram estruturas químicas, removeram medições duvidosas e focaram em compostos que compartilham um grupo quelante de zinco comum a essa classe de inibidores. Usando limites rígidos, eles rotularam moléculas como claramente ativas ou claramente inativas e descartaram casos limítrofes que poderiam confundir os modelos. Como havia muito mais moléculas inativas do que ativas, balancearam os dados para que os algoritmos de aprendizado não passassem a favorecer simplesmente a classe majoritária. Também usaram uma divisão baseada em “scaffolds” (andaimes estruturais) para que conjuntos de treino e teste contivessem núcleos moleculares diferentes, oferecendo uma visão mais realista de quão bem os modelos lidariam com compostos verdadeiramente novos.
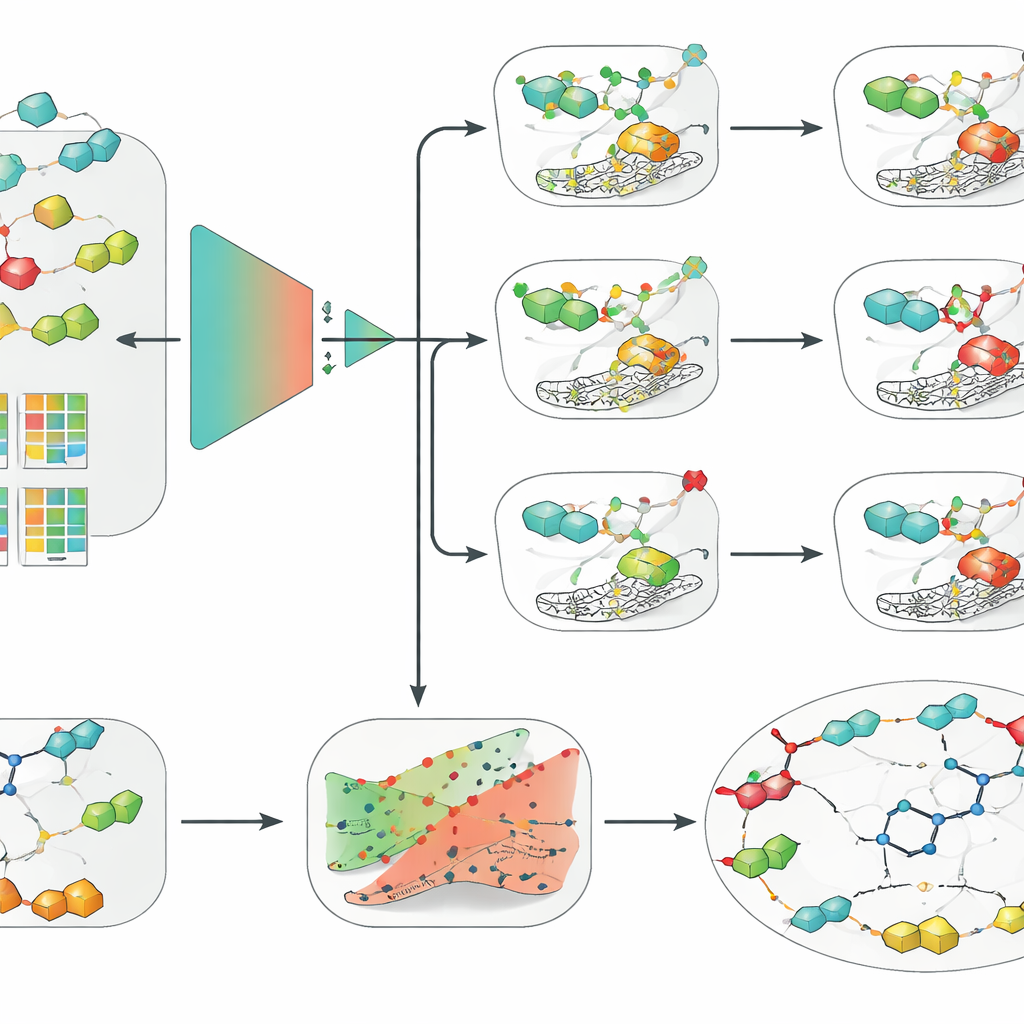
Modelos simples superam deep learning quando os dados são limitados
Com esse conjunto de dados curado, a equipe comparou uma ampla gama de abordagens, desde métodos clássicos de aprendizagem de máquina como regressão logística, florestas aleatórias e máquinas de vetores de suporte (SVMs) até redes neurais profundas modernas, incluindo modelos baseados em grafos que operam diretamente sobre estruturas moleculares. Eles combinaram esses métodos com várias formas de codificar moléculas, como descritores tradicionais feitos à mão, fingerprints baseadas em chaves e embeddings aprendidos de um modelo de linguagem química. Em todas as três isoformas da enzima e sob a avaliação mais rígida baseada em scaffolds, uma combinação destacou‑se consistentemente: uma SVM alimentada com fingerprints de conectividade estendida, uma forma estruturada de descrever ambientes químicos locais dentro de uma molécula. Surpreendentemente, essa configuração comparativamente simples superou modelos de grafos e deep learning mais em voga, ressaltando que qualidade dos dados, validação cuidadosa e bons descritores moleculares podem importar mais que a complexidade algorítmica quando os conjuntos de dados são de tamanho moderado.
Adicionando confiança confiável e explicações acessíveis
Os pesquisadores então envolveram seu melhor modelo SVM em duas camadas adicionais projetadas para tornar suas predições mais utilizáveis na descoberta de fármacos. Primeiro, aplicaram uma estrutura chamada predição conformal, que não só fornece uma resposta sim‑ou‑não, mas entrega uma região de resultados prováveis junto com uma taxa de erro garantida. Isso permite que os cientistas ajustem quão cauteloso querem que o modelo seja e reconheçam casos em que o modelo está genuinamente incerto. Em segundo lugar, usaram explicações contrafactuais para tornar o raciocínio do modelo mais intuitivo. Para uma dada molécula, geraram análogos estreitamente relacionados que invertem o resultado previsto de ativo para inativo, ou vice‑versa. Examinando esses pares para o candidato clínico SLC‑0111, que bloqueia seletivamente IX e XII mas não II, o método redescobriu independentemente um insight importante da química medicinal: pequenas mudanças na “cauda” da molécula alteram fortemente qual isoforma ela prefere se ligar.
Dos algoritmos a ferramentas práticas de desenho de fármacos
Para tornar sua abordagem acessível, os autores empacotaram os três modelos SVM, a camada de incerteza e o mecanismo contrafactual em uma ferramenta gráfica chamada CAInsight. Um usuário pode fornecer a representação textual de uma molécula e, com um único clique, obter a atividade prevista contra hCA II, IX e XII, uma estimativa de quão confiável é cada predição e sugestões de ajustes estruturais que podem aumentar ou reduzir a atividade. Embora os modelos se concentrem em classificar moléculas como ativas ou inativas em vez de prever potência exata ou seletividade em um único passo, eles já reproduzem comportamentos conhecidos para candidatos reais a fármacos e distinguem mudanças estruturais sutis. Os autores observam que conjuntos de dados maiores e mais uniformes, além de uma análise mais profunda de como os limites de atividade são escolhidos, poderiam refinar ainda mais o desempenho.
O que isso significa para futuros medicamentos contra o câncer
Em termos simples, este trabalho mostra que modelos de aprendizagem de máquina cuidadosamente construídos e bem explicados podem ajudar químicos a projetar medicamentos contra o câncer que distingam melhor entre alvos enzimáticos muito semelhantes. Ao combinar estatísticas robustas, estimativas de incerteza e exemplos intuitivos de “e se”, a estrutura não só prevê quais moléculas têm maior probabilidade de funcionar, mas também sugere por quê. Esse tipo de inteligência artificial transparente pode acelerar triagens virtuais, apoiar o desenho gerativo de novos compostos e reduzir o esforço de tentativa e erro no laboratório, auxiliando em última instância a descoberta de tratamentos mais seletivos e seguros para pacientes.
Citação: Ghamsary, M.S., Rayka, M. & Naghavi, S.S. Interpretable machine learning rationalizes carbonic anhydrase inhibition via conformal and counterfactual prediction. Sci Rep 16, 8419 (2026). https://doi.org/10.1038/s41598-026-39771-2
Palavras-chave: inibidores da anidrase carbônica, aprendizagem de máquina interpretável, seletividade de fármacos, predição conformal, explicações contrafactuais