Clear Sky Science · pt
Mutações composês heterozigóticas no gene CHAT, uma missense e uma variante no sítio de splicing, em dois irmãos com síndrome miastenica congênita
Quando a respiração falha sem aviso
Algumas crianças parecem saudáveis ao nascer, mas de repente param de respirar durante febres leves, necessitando de ventilação de emergência. Para suas famílias, os episódios são aterrorizantes e misteriosos. Este estudo investiga dois desses irmãos do Japão e rastreia seus ataques de fraqueza e apneia (pausas na respiração) até pequenas alterações em um único gene que ajuda os nervos a se comunicarem com os músculos. Ao juntar pistas clínicas, sequenciamento gênico e modelagem de proteínas por computador, os pesquisadores mostram como essas mutações provavelmente comprometem uma enzima-chave e oferecem aos médicos um alvo mais claro para diagnóstico e tratamento.
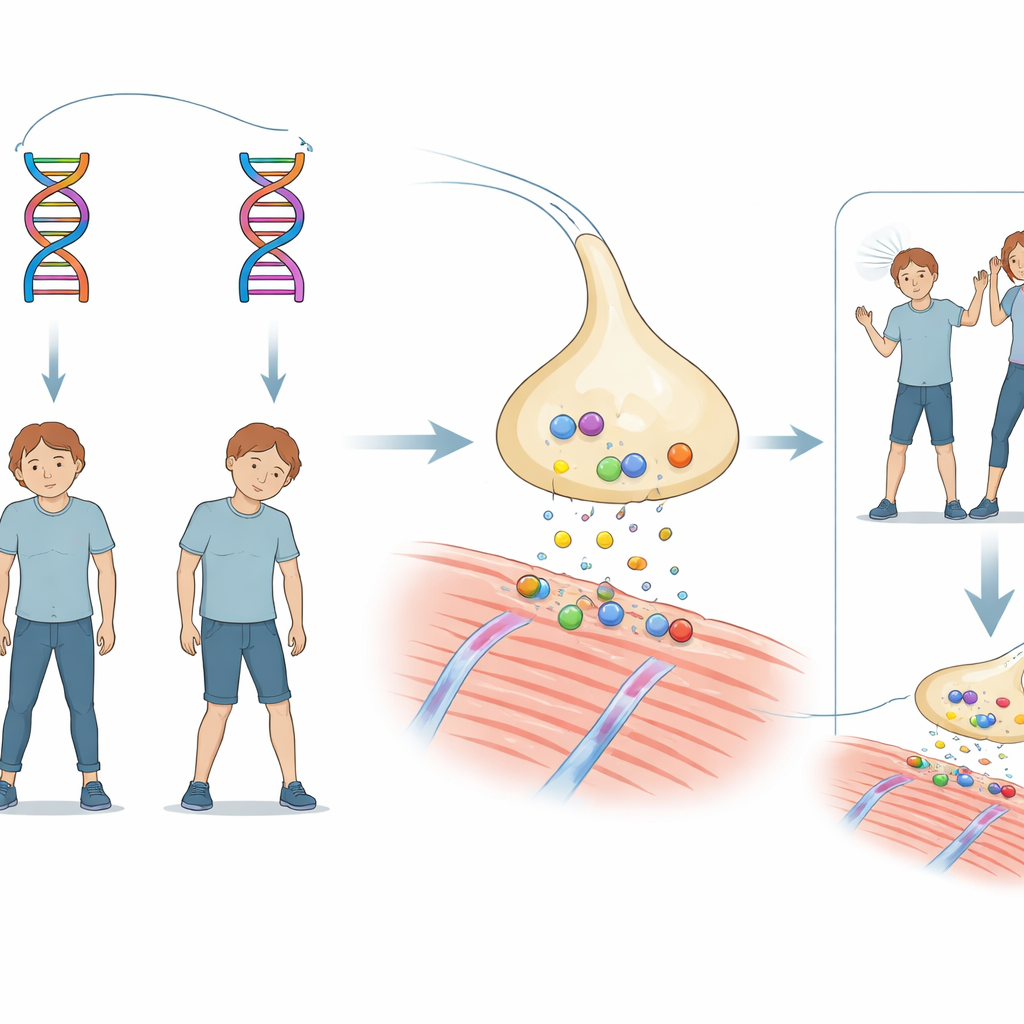
Um mistério familiar de fraqueza súbita
A história centra-se em um irmão e em uma irmã que ambos apresentaram desenvolvimento motor ligeiramente lento na primeira infancia. Por volta dos 18 meses de idade, cada um teve episodios de apneia e perda de conscientência durante febres, graves o suficiente para requerer ventilação. Ao crescerem, ambos continuaram a ter crises de ptose (pálpebras caídas) e fraqueza muscular generalizada desencadeadas por infecções, febres ou esforço. Tomografias cerebrais foram normais, e formas comuns de miastenia mediadas por anticorpos (uma doença em que a comunicação entre nervo e músculo é prejudicada) foram excluídas. No entanto, um fármaco que aumenta o sinal quı́mico entre nervos e músculos melhorou claramente seus sintomas, apontando para uma condição hereditária rara chamada síndrome miastênica congênita.
Encontrando as instruções defeituosas
Para procurar uma causa hereditária, a equipe sequenciou todos os genes codificadores de proteínas dos irmãos e de seus pais. Eles descobriram que cada criança carregava duas alterações diferentes no mesmo gene, CHAT, que codifica a colina acetiltransferase — uma enzima que produz acetilcolina, o principal mensageiro quı́mico usado pelos nervos para ativar os músculos. Uma alteração trocou um único bloco de construção da enzima (uma mutação missense conhecida como G411R). A outra situava-se em uma fronteira crı́tica onde a célula normalmente corta e junta segmentos do gene durante a produção de RNA (uma mutação no sítio de splicing rotulada c.752+2T>C). Cada pai carregava apenas uma dessas mudanças e era saudável; somente as crianças que herdaram ambas apresentaram a doença, sugerindo que o par de mutações em conjunto enfraquece a função da enzima.
Investigando como um corte oculto altera a enzima
Como os pesquisadores não conseguiram obter RNA CHAT natural suficiente a partir de células sanguı́neas, usaram um experimento de "minigene". Eles clonaram o trecho relevante do gene em um vetor de DNA, introduziram a versão normal ou a mutada em células em cultura e então examinaram como o RNA foi processado. No construto normal, o RNA continha todos os segmentos esperados. Na versão mutante, um segmento inteiro conhecido como exon 5 foi omitido, embora o quadro de leitura global do gene tenha permanecido intacto. Isso significava que a enzima seria sintetizada, porém faltando um curto trecho interno de aminoácidos. Comparações evolutivas mostraram que essa região ausente é altamente conservada entre espécies, sugerindo que desempenha um papel estrutural importante.
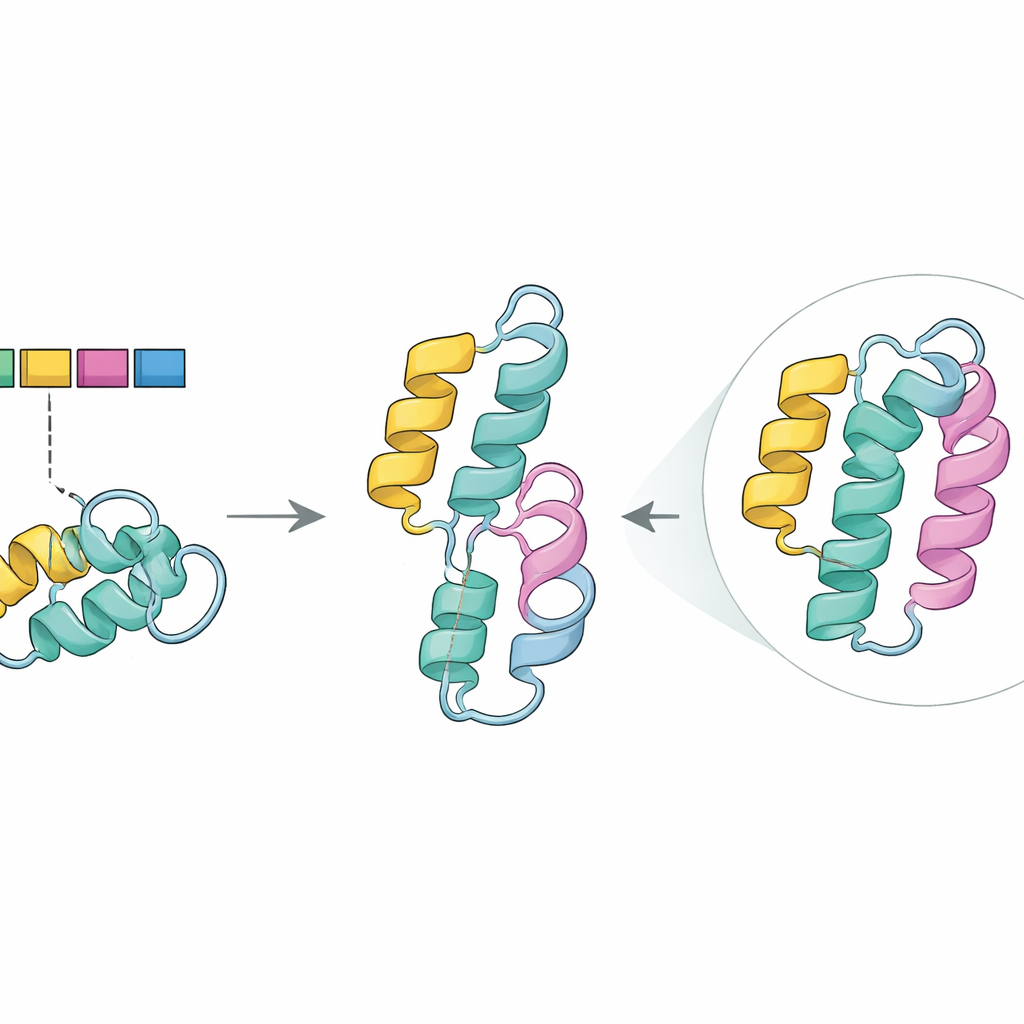
Vendo danos estruturais in silico
Para explorar esse papel, a equipe recorreu ao AlphaFold2, um programa avançado que prediz as formas tridimensionais de proteı́nas a partir de suas sequências. Na enzima normal, a porção codificada pelo exon 5 forma um dos segmentos em espiral fortemente compactados (uma alfa-hélice) que ajudam a estabilizar o núcleo da proteı́na. Na estrutura mutante prevista, essa hélice desapareceu, deixando uma lacuna em uma região conhecida por trabalhos anteriores como crucial para manter a estabilidade e favorecer uma quı́mica eficiente. Juntamente com ferramentas computacionais que identificam mutações danosas, esses resultados apoiam a ideia de que a omissão do exon 5, especialmente quando emparelhada com a mudança G411R na outra cópia do gene, compromete o desempenho da enzima sem eliminá‑la completamente — consistente com os sintomas moderados, mas graves, observados nos irmãos.
O que isso significa para pacientes e famílias
O estudo conclui que a combinação da mutação missense G411R e da recém-identificada mutação no sítio de splicing no CHAT é muito provavelmente responsável pela síndrome miastênica congênita dos irmãos. Ao demonstrar, por meio do ensaio de minigene e da modelagem estrutural, como a alteração no sítio de splicing remove uma hélice estabilizadora da enzima, os autores fornecem uma explicação mecanı́stica sobre a qual clínicos e pesquisadores podem se apoiar. Para famílias afetadas, esse trabalho oferece mais do que um nome: apoia tratamentos personalizados com fármacos que aumentam a sinalização neuromuscular, orienta o aconselhamento genético para gestações futuras e acrescenta um exemplo importante ao catálogo de como mudanças sutis em nosso código genético podem influenciar profundamente a força muscular e o ato básico de respirar.
Citação: Kikuchi, S., Wada, N., Mariya, T. et al. Compound heterozygous CHAT gene mutations, a missense and a splice site variant, in two siblings with congenital myasthenic syndrome. Sci Rep 16, 9346 (2026). https://doi.org/10.1038/s41598-026-39759-y
Palavras-chave: síndrome miastênica congênita, gene CHAT, colina acetiltransferase, mutação no sítio de splicing, junção neuromuscular