Clear Sky Science · pt
Deconvolução aprimorada guiada por atenção possibilita estimativa de tipos celulares sem referência em transcriptômica espacial
Vendo células no seu lugar
A biologia moderna pode ler a atividade de milhares de genes de uma só vez, não apenas em células isoladas, mas diretamente em fatias finas de tecido. Essa visão de “transcriptômica espacial” revela onde diferentes células vivem e interagem, mas cada medição frequentemente mistura sinais de muitas células vizinhas. O estudo apresenta um novo método computacional, chamado AGED, que pode desembaraçar essas misturas e estimar quais tipos celulares estão presentes em cada local — sem precisar de um conjunto de referência de célula única separado e cuidadosamente correspondente. 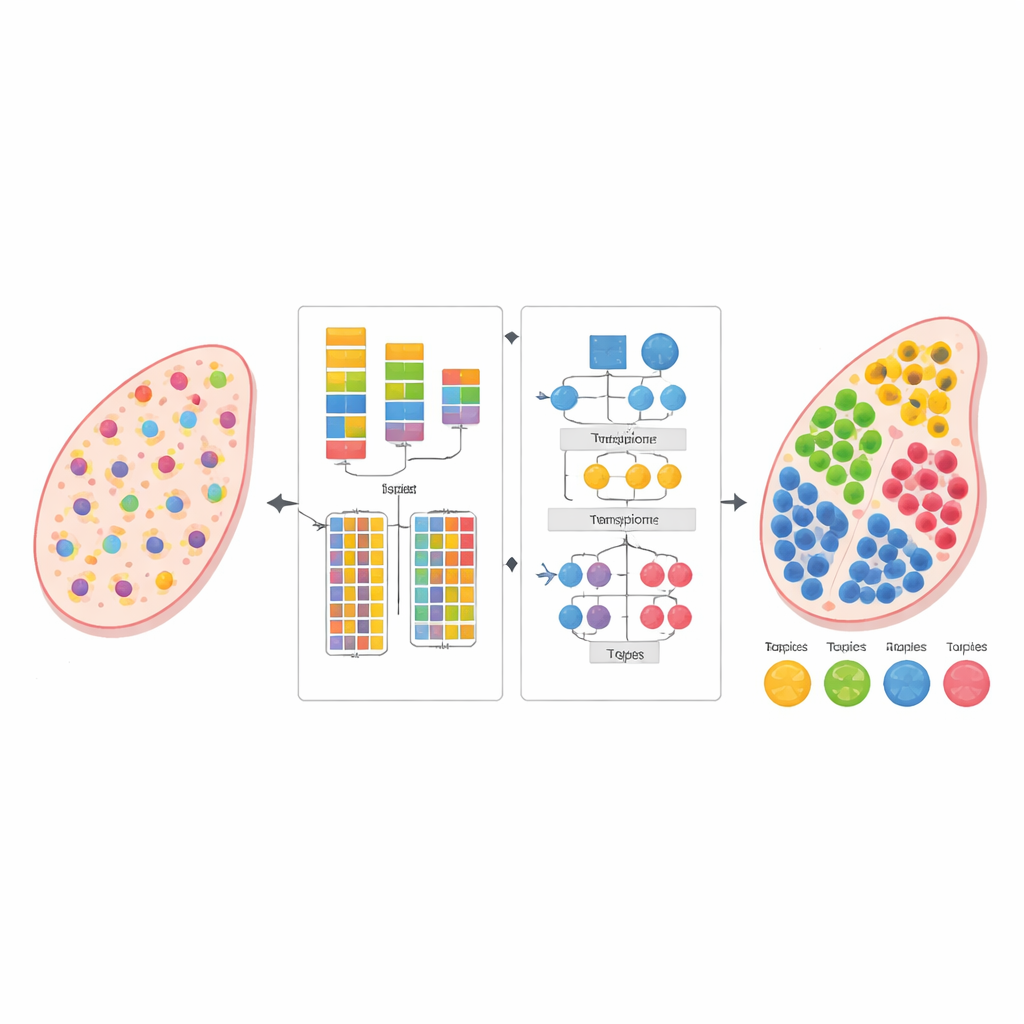
Por que mapear células em tecidos é difícil
Plataformas de transcriptômica espacial medem a atividade gênica através de uma grade de pontos sobre uma fatia de tecido. Como a maioria desses pontos captura várias células ao mesmo tempo, os pesquisadores precisam decompor matematicamente os sinais mistos para recuperar os tipos celulares subjacentes e suas proporções. Ferramentas existentes frequentemente dependem de atlas de referência de célula única externos do mesmo tecido. Esses atlas podem faltar para tecidos raros, estados patológicos especiais ou condições experimentais incomuns e, mesmo quando disponíveis, podem não corresponder perfeitamente, introduzindo vieses. Métodos sem referência evitam essa dependência, mas as abordagens atuais têm dificuldade com padrões espaciais complexos, relações genéticas sutis e o desafio de decidir quantos tipos celulares distintos procurar em primeiro lugar.
Uma estratégia em dois passos para desembaraçar misturas
Os autores projetaram o AGED como uma estrutura em duas etapas que combina ideias de estatística e aprendizado profundo moderno. Na primeira etapa, o método testa uma gama de possibilidades sobre quantos tipos celulares podem estar presentes no tecido. Ele usa uma rede neural baseada em atenção rápida, conhecida como Performer, para aprender decomposições candidatas e então avaliá-las usando vários critérios ao mesmo tempo: quão bem o modelo reconstrói as contagens gênicas observadas, quão claramente os grupos celulares inferidos se separam uns dos outros e quão diversos esses grupos são. Um procedimento de ajuste de curva encontra um “ponto de cotovelo” onde adicionar mais tipos celulares traz pouco benefício, permitindo que o método selecione automaticamente um número adequado em vez de depender do palpite do usuário.
Atenção guiada para capturar a biologia
Uma vez definido o número de tipos celulares, a segunda etapa do AGED refina a solução com uma arquitetura de atenção mais rica. Ela parte de um modelo estatístico de tópicos que trata cada ponto do tecido como uma mistura de “temas” ocultos — aqui representando tipos celulares — e cada tipo celular como um padrão gênico característico. Esses temas iniciais fornecem uma estrutura global. O modelo então sobrepõe vários mecanismos de atenção: um conecta os temas estatísticos à rede neural, outro agrega informações de pontos vizinhos no espaço físico, e um terceiro liga temas diretamente aos genes. Um sistema de gating permite que o modelo decida, para cada caso, quanto confiar nos padrões estatísticos prévios versus os dados locais. Restrições adicionais incentivam soluções esparsas, refletindo a realidade biológica de que a maioria das localidades do tecido é dominada por apenas alguns tipos celulares principais. 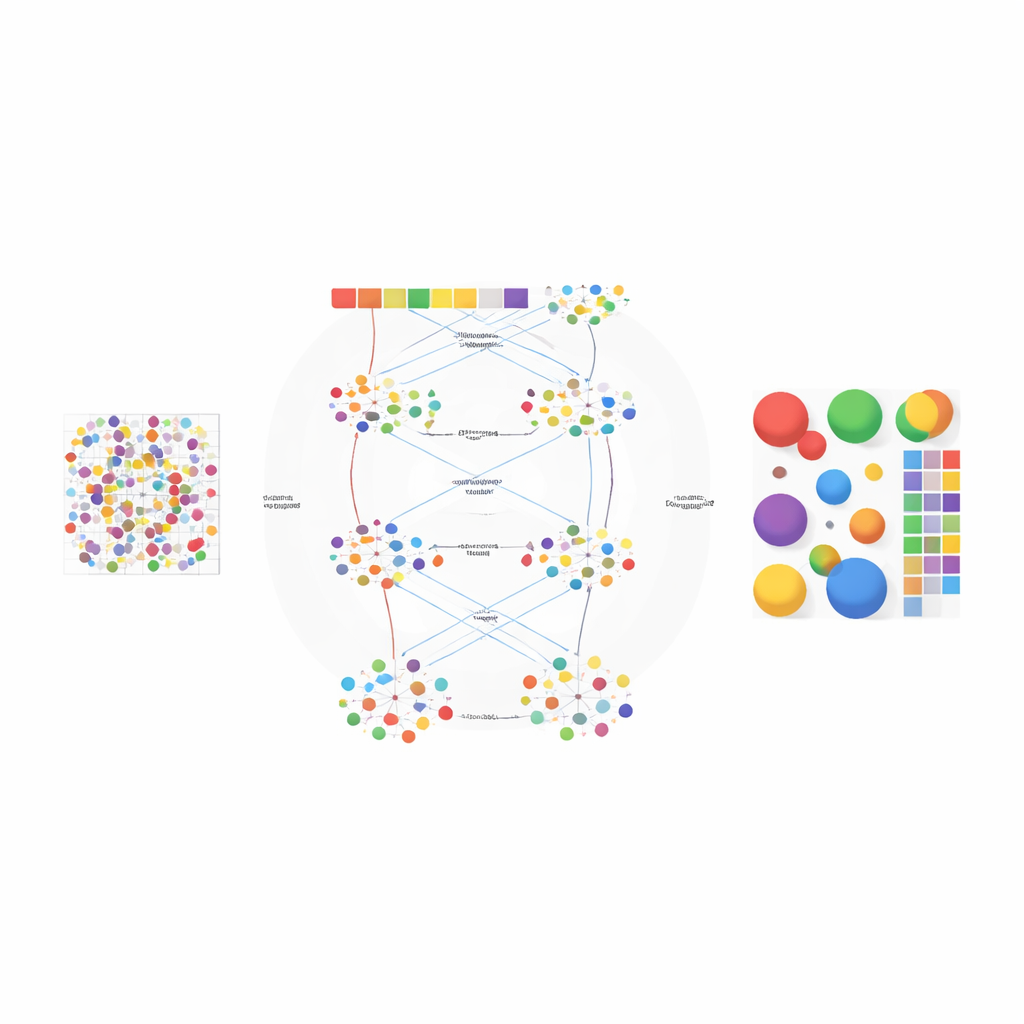
Colocando o método à prova
Os pesquisadores avaliaram o AGED em vários tipos de dados. Em tecido simulado do bulbo olfatório de camundongo, o método recuperou quatro camadas anatômicas conhecidas e correspondeu às composições celulares reais mais de perto do que ferramentas amplamente usadas com e sem referência, alcançando alta correlação com a verdade terreno e baixo erro de reconstrução. Em adenocarcinoma ductal pancreático humano, o AGED escolheu automaticamente uma solução de vinte tipos celulares que se alinhou com regiões anotadas por patologistas, como tumor, ducto e pâncreas normal, superando outros métodos em uma medida de similaridade estrutural que compara mapas inferidos com a estrutura tecidual visível. Em tecido de timo humano, o AGED separou com precisão populações celulares chave e capturou uma relação biologicamente esperada negativa entre dois tipos epiteliais especializados — um padrão que abordagens concorrentes não conseguiram reproduzir. Análises adicionais em outros conjuntos de dados e em resolução semelhante à de célula única apoiaram ainda mais a robustez do método.
O que isso significa para o futuro
Para um público não especialista, o AGED pode ser visto como um motor inteligente de desmistura para tecidos complexos: ele aprende quantas comunidades celulares distintas existem, onde estão localizadas e quais genes as definem, tudo a partir dos próprios dados espaciais. Ao tecer modelos estatísticos interpretáveis com redes neurais flexíveis baseadas em atenção, a estrutura oferece precisão e insight, mesmo quando não existe um atlas de referência adequado. Isso a torna uma ferramenta prática para explorar a organização tecidual em saúde e doença, de camadas cerebrais a tumores e órgãos imunológicos, e aponta para uma estratégia mais ampla de usar conhecimento prévio para guiar modelos de aprendizado de máquina poderosos, porém opacos, na biologia.
Citação: Yang, X., Wang, Y. & Chen, X. Attention-guided enhanced deconvolution enables reference-free cell type estimation in spatial transcriptomics. Sci Rep 16, 8097 (2026). https://doi.org/10.1038/s41598-026-39703-0
Palavras-chave: transcriptômica espacial, deconvolução de tipos celulares, aprendizado profundo, arquitetura tecidual, análise sem referência