Clear Sky Science · pt
Modelagem Matemática e Cálculo de Índices NM‑Polinomiais para Predição de Propriedades Físico‑químicas
Por que isso importa para futuros medicamentos
Projetar um novo medicamento é um pouco como projetar uma aeronave: você quer saber como ele se comportará muito antes de construí‑lo de fato. Para fármacos, esse comportamento inclui quão facilmente evaporam, como se misturam com água ou lipídios e como se movem pelo corpo. Este artigo mostra como matemática cuidadosamente elaborada pode prever muitas dessas características físicas e químicas a partir apenas da estrutura de um fármaco, potencialmente poupando tempo, custo e tentativas e erros na descoberta de medicamentos. 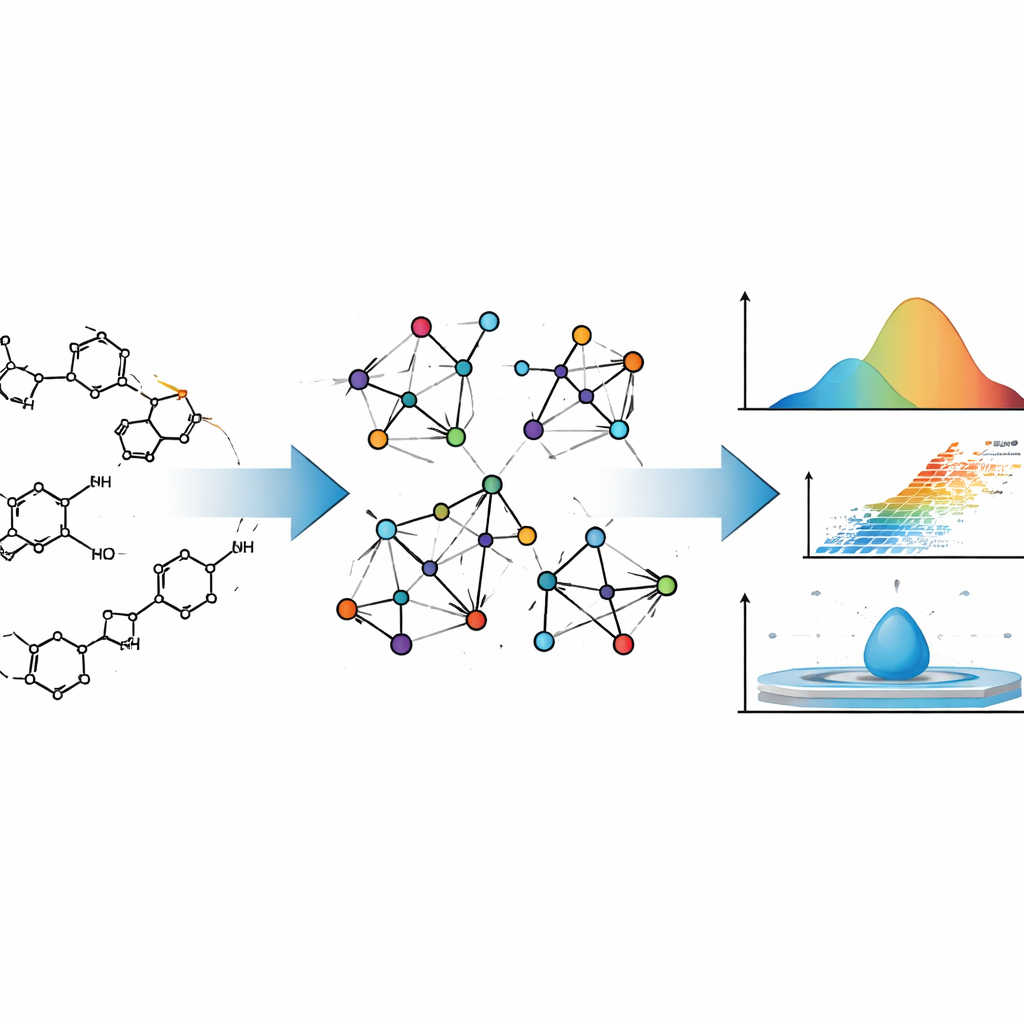
De moléculas a redes
Os autores tratam moléculas de fármacos não apenas como coleções de átomos, mas como redes. Nessa visão, cada átomo é um ponto e cada ligação química é uma linha que conecta dois pontos. Esse tipo de descrição vem da teoria dos grafos, um ramo da matemática que estuda redes de todos os tipos, desde conexões em redes sociais até redes elétricas. Químicos usam tais “grafos moleculares” há décadas, porque certos resumos numéricos desses grafos — chamados índices topológicos — frequentemente acompanham o comportamento real das moléculas, como facilidade de ebulição ou densidade.
Adicionando detalhe de vizinhança à imagem
Índices tradicionais geralmente prestam atenção apenas em quantas ligações tocam cada átomo. A equipe por trás deste estudo vai um passo além. Eles usam os chamados índices NM‑polinomiais (neighborhood M‑polynomial), que não apenas contam as próprias conexões de um átomo, mas também resumem quão conectados são seus vizinhos. Essa descrição mais rica captura sutilezas como o grau de ramificação de uma molécula, como seus anéis estão fundidos e onde átomos de oxigênio ou nitrogênio se situam na estrutura. Essas características, por sua vez, influenciam o quanto as moléculas se aderem umas às outras, quão rígidas são e como seus elétrons respondem a campos elétricos — todos ingredientes de propriedades físico‑químicas importantes.
Testando a ideia em fármacos contra o câncer reais
Para ancorar a matemática na realidade, os autores primeiro calculam índices NM‑polinomiais para dois agentes antineoplásicos bem conhecidos, Mitoxantrona e Doxorrubicina. Ambos são moléculas complexas com múltiplos anéis, amplamente usadas em quimioterapia. Ao traduzir seus desenhos químicos detalhados em grafos moleculares e depois em índices NM‑polinomiais, os autores mostram como o método acompanha sistematicamente mudanças estruturais em diferentes “tamanhos” dessas moléculas. Em seguida, automatizam esse processo com um programa em Python, que recebe a conectividade da molécula (na forma de uma matriz de adjacência) e retorna instantaneamente o conjunto completo de índices, minimizando erro humano e acelerando cálculos que seriam tediosos manualmente. 
Treinando máquinas para ler impressões digitais moleculares
Em seguida, os pesquisadores ampliam além desses dois fármacos para uma coleção mais ampla de 45 medicamentos policíclicos, incluindo nomes comuns como paracetamol, ibuprofeno e várias terapias-alvo modernas. Para cada fármaco, compilam nove índices NM‑polinomiais e nove propriedades medidas experimentalmente: complexidade, ponto de ebulição, entalpia de vaporização, ponto de fulgor, refratividade molar, polarizabilidade, tensão superficial, volume molar e índice de refração. Depois treinam vários modelos de regressão ao estilo de aprendizado de máquina — Linear, Ridge, Lasso e Elastic Net — para aprender como combinações de índices se relacionam com cada propriedade. Salvaguardas estatísticas cuidadosas são usadas ao longo do processo: removem entradas redundantes, padronizam variáveis, realizam validação cruzada repetida em 80% dos dados e testam os modelos finais em 20% não utilizados.
O que os números revelam
Os modelos mostram que os índices NM‑polinomiais são particularmente poderosos para propriedades ligadas a como as moléculas se empacotam e interagem. Para ponto de ebulição, entalpia de vaporização, ponto de fulgor, refratividade molar, polarizabilidade e volume molar, os melhores modelos atingem pontuações de correlação muito altas, o que significa que os valores previstos acompanham de perto os experimentais. Métodos regularizados como Ridge e Elastic Net geralmente apresentam melhor desempenho, sugerindo que impor restrições suaves ajuda os modelos a focarem nos aspectos mais informativos dos índices. Um mapa de correlação confirma que vários índices — especialmente os relacionados à conectividade geral e à “riqueza de vizinhança” — estão fortemente e consistentemente alinhados com essas propriedades ao longo do painel de 45 fármacos.
Limites e espaço para melhoria
Nem toda propriedade coopera. O índice de refração, que captura como a luz se curva ao entrar em um material, prova‑se teimoso: os modelos têm dificuldade em superar médias simples, e os índices NM‑polinomiais mostram apenas correlações fracas com ele. A tensão superficial é capturada de forma moderada, mas não tão fortemente quanto as outras características. Essas lacunas indicam que alguns comportamentos dependem de características além da conectividade bidimensional, como forma tridimensional ou efeitos eletrônicos sutis. Os autores sugerem que trabalhos futuros poderiam combinar índices NM‑polinomiais com descritores quântico‑químicos ou 3D para preencher essa lacuna.
O que isso significa para o desenho de fármacos
Em termos simples, o estudo mostra que matemática sofisticada, porém bem estruturada, pode transformar um esboço estático de uma molécula em um preditor surpreendentemente preciso de como essa molécula se comporta em laboratório. Para muitas propriedades importantes — quão difícil é ferver, quão volumosa é ou com que facilidade seus elétrons se deslocam — a abordagem NM‑polinomial, combinada com técnicas de regressão modernas, iguala ou supera métodos anteriores que usavam índices mais simples ou conjuntos de dados menores. Embora ainda não substitua por completo experimentos, oferece aos projetistas de medicamentos uma ferramenta de triagem mais rápida: calculando essas impressões digitais baseadas em grafos, eles podem estimar propriedades físico‑químicas chave desde cedo, concentrar o trabalho de laboratório nos candidatos mais promissores e explorar o espaço químico com mais eficiência.
Citação: Tawhari, Q.M., Naeem, M., Koam, A.N.A. et al. Mathematical Modeling and Computation of NM-Polynomial Indices for Physicochemical Properties Prediction. Sci Rep 16, 8136 (2026). https://doi.org/10.1038/s41598-026-39562-9
Palavras-chave: teoria dos grafos químicos, predição de propriedades de fármacos, topologia molecular, aprendizado de máquina em química, descritores físico‑químicos